
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis of a helicene-fused porphyrin leading to a π-extended chiral chromophore†
Vincent
Silber
a,
Nathalie
Gruber
b,
Marion
Jean
c,
Nicolas
Vanthuyne
c and
Romain
Ruppert
 *a
*a
aInstitut de Chimie, UMR CNRS 7177, Institut Le Bel, Université de Strasbourg, 4 rue Blaise Pascal, 67000, Strasbourg, France. E-mail: rruppert@unistra.fr
bFédération de Chimie Le Bel, FR 2010, BP 296R8, 1 rue Blaise Pascal, 67008, Strasbourg Cedex, France
cAix Marseille Univ, UMR CNRS 7313, Centrale Marseille, iSm2, 13397, Marseille cedex 20, France
First published on 26th April 2022
Abstract
The preparation of several covalently linked [6]-helicene-porphyrins is reported. A fused [6]-helicene-porphyrin π-extended aromatic system was isolated, the enantiomers separated and the chiroptical properties determined. The oxidative cyclodehydrogenation proved to be very effective for six-membered fused helical systems, but not suited for the formation of five-membered fused systems.
The syntheses of polycyclic (hetero)-aromatic hydrocarbons (PAHs) have been extensively developed recently because new synthetic methods and better analytical tools have given easier access to these large molecules.1,2 These molecules were prepared to provide experimental data to better understand the π-electron distribution in these molecules. Unusual physical or mechanical properties were also expected for these small molecular analogues of graphene. Among this class of compounds, distorted large aromatic molecules are potentially chiral compounds.3,4ortho-Fused polycyclic aromatic hydrocarbons, named helicenes, are chiral PAHs and their chemistry has been developed in the last two decades due to improved synthetic access.5 Their intrinsic chirality might lead to applications in material sciences, asymmetric synthesis or catalysis. In particular, chiroptical properties are expected from enantiopure thermally stable helicenes. The two enantiomers of helicenes are stable as soon as the number of ortho-fused rings is at least equal to six.6 Light absorbance of simple helicenes occurs mainly in the UV region, but incorporating a chromophore will generate a chiral compound also able to absorb visible light. Porphyrinoids, the pigments of life, are large aromatic systems absorbing in the visible region. The presence of metal ions in their cores brings additional physical or chemical properties compared to other polyaromatics. Chiral porphyrinoids are known, but examples of porphyrins with intrinsic chirality are much rarer.7 Helical porphyrinoids have been prepared and for some of them both enantiomers were separated and their chiroptical properties studied.
Extended porphyrins present very interesting electronic properties and their synthesis was improved markedly during the last two decades. Fusing other aromatics to the porphyrin core has led to chromophores absorbing light in the visible and the near-infrared regions.8 Among other synthetic routes to extended porphyrins, the cyclodehydrogenative oxidation (Scholl reaction) has provided access to many new porphyrinoids.9 Very often, one or several meso-substituents were fused to the porphyrin core via a five or six-membered ring (see Fig. 1).
 | ||
| Fig. 1 Five- vs. six-membered fused meso-aryl groups (R, R1, and R2 electron-donating substituents). | ||
Herein, we report the synthesis and structural, optical, electrochemical and circular dichroism properties of porphyrin–helicene scaffolds. In particular, the preparation, the electronic properties, the separation of the enantiomers, and the electronic circular dichroism of a fused porphyrin-helicene compound are described.
The Scholl reaction conditions were applied to compound 1, a tetraarylporphyrin bearing three meso-aryl groups substituted twice in their ortho positions and one meso-aryl group with two electron donating methoxy substituents (see Scheme 1). With this substitution pattern, the formation of only one C–C bond formation remained possible. A green compound 2 was isolated in 60% yield when iron(III) chloride was used as an oxidant.
 | ||
| Scheme 1 Oxidation of nickel(II)porphyrin 1. | ||
After this encouraging result with a simple nickel(II) porphyrin, a nickel(II) porphyrin bearing a helicene as meso-aryl group was prepared. The enantiomers of [6]-helicenes are known to be thermally stable6 and two methoxy groups were added to obtain an electron rich meso-aryl group. These new functionalised [6]-helicenes were prepared according to the well-known 6π-electrocyclisation followed by aromatisation.10 Reacting the aldehyde derivative of this dimethoxy-[6]-helicene, three equivalents of 2,6-dimethyl-4-tert-butyl-benzaldehyde, with pyrrole under classical Lindsey condition gave, after chromatographic separations and in acceptable yield, the desired tetraaryl porphyrin base 3 (see Scheme 2). Metalation with Ni(acac)2 afforded in almost quantitative yield nickel(II) porphyrin 4.
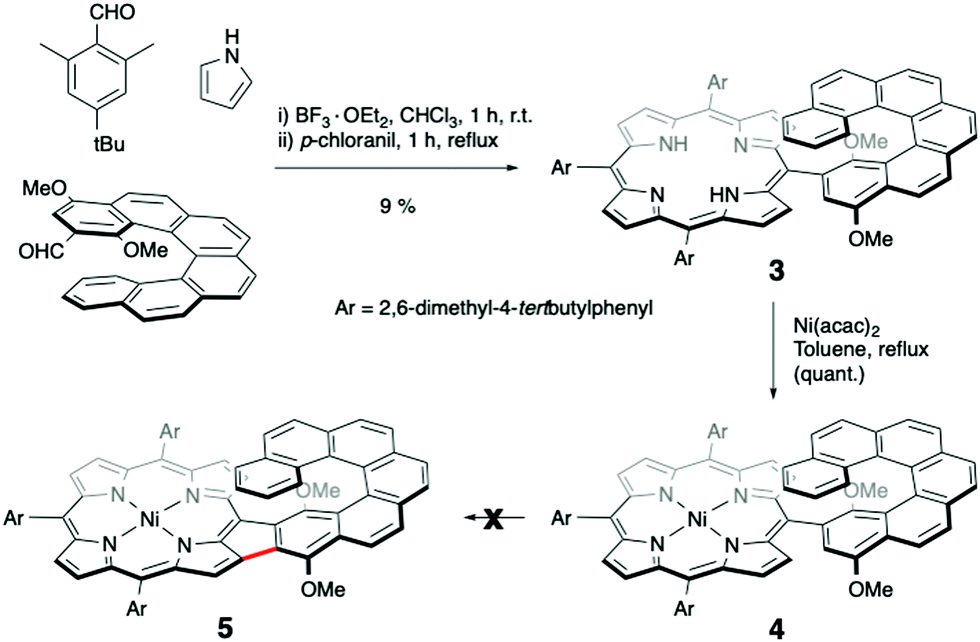 | ||
| Scheme 2 Synthesis of a nickel(II)porphyrin bearing a [6]-helicenic meso-substituent. | ||
Again, it should be noted that only one C–H bond (labelled C22 in Fig. 2) of the first aromatic ring of the helicenic part was available for the envisaged Scholl reaction. This free base porphyrin was characterised by standard techniques (NMR and MS) and single crystals suitable for X-ray were obtained (see Fig. 2).
 | ||
| Fig. 2 X-Ray structure of free base porphyrin 3 (three meso-aryl groups omitted for clarity). | ||
Despite many attempts to realise a cyclodehydrogenation reaction with nickel(II) porphyrin 4, the fused porphyrin-helicene 5 was never isolated. Many reported Scholl reaction conditions with numerous oxidants (Fe(III), DDQ/TfOH, DDQ/Sc(OTf)3, PIFA, …) were tried, but without success.
Several green products were obtained as traces after reaction with iron(III) chloride, but always as a complicated mixture of polychlorinated products in addition to degradation products. This result was quite surprising, because the intramolecular oxidative aromatic coupling gave cleanly compound 2. From an electronic point of view, compounds 4 and 1 are almost identical and therefore it seemed that steric hindrance was preventing the oxidative coupling in the case of porphyrin 4.
The alternative approach to the six-membered oxidative fusion was then attempted. Another substitution pattern was needed for the [6]-helicenic part (see Scheme 3) and the desired compound was obtained by a similar reaction scheme, but with different substitutions (see ESI† for experimental details).
 | ||
| Scheme 3 Comparison between two substitution patterns for the [6]-helicenic aldehydes used and available positions for the oxidative cyclisation highlighted in colour. The starting aldehyde was prepared according to published procedures.10 | ||
The free base porphyrin 6 was again obtained by the statistical Lindsey conditions in 11% yield (see Scheme 4). Quantitative metalation with Ni(acac)2 afforded the nickel(II)porphyrin 7. At this point, it should be emphasised that in this new approach, two cyclisations were possible leading to five- or six-membered rings after oxidation (positions respectively highlighted in orange and blue in Scheme 3). Despite this, only one cyclisation occurred in the presence of iron(III) chloride and the six-membered fused compound 8 was isolated in 89% yield after chromatographic purification and crystallisation.
 | ||
| Scheme 4 Synthesis of nickel(II)porphyrin 8 fused with a [6]helicene. | ||
The success of the cyclisation could be easily visualised, with the orange colour of starting compound 7 changing to olive-green after oxidation, affording product 8 (see Fig. 3). The high-resolution mass spectra clearly showed the loss of two hydrogen atoms with the mass diminishing from 1202.53 (exact mass for 7: C83H76N4NiO) to 1200.52 (exact mass for 8: C83H74N4NiO). The position of cyclisation (blue vs. orange in Scheme 3) was determined by 1H NMR. If the cyclisation involves the blue position of the helicenic subunit, the two protons next to the methoxy-substituent will appear as two doublets (indeed observed at 6.51 and 7.89 ppm, see ESI†). The other possible cyclisation would afford a singlet for the proton next to the methoxy-substituent.
 | ||
| Fig. 3 Electronic spectra of compound 7 (orange line) and of fused compound 8 (green line). | ||
Preferences for five- or six-membered ring formation during the Scholl oxidation were observed earlier.11 The group of Osuka checked the reaction of dibenzo[a,g]corannulene with an M(II) porphyrin (M = Zn or Ni). When this large aromatic substituent was located at the meso-position, the oxidation reaction leading to the five-membered fused ring was observed. Steric hindrance for the formation of the six-membered ring was invoked to explain this high regioselectivity.12 The formation of the six-membered ring to form compound 8 during the Scholl reaction kept the helicene far away from the porphyrin ring, whereas the cyclisation to form the five-membered ring would bring the helicenic moiety much closer to the porphyrin ring. The unsuccessful attempts to cyclise nickel(II) porphyrin 4 clearly illustrated this point.
The electronic spectra of the starting nickel(II) porphyrin 7 and the fused product 8 are represented in Fig. 3. The expected bathochromic shift after cyclisation and extension of the aromatic core was observed for the Soret and the Q-bands. The formation of the six-membered fused connection increased also the absorbance of the Q-bands compared to the absorbance of the Q-bands for the five-membered fused compound 2. The lowest energy band was shifted from 530 nm for 7 to 662 nm for 8, reflecting the large decrease of the optical gap (from 2.34 eV to 1.87 eV). The same lowering of the HOMO–LUMO gap was found during electrochemical measurements, the gap between the first oxidation and the first reduction decreased from 2.42 V to 1.93 V (see Fig. 4). The two reversible oxidation and the two reduction waves of compound 8 are typical of nickel(II) porphyrins (all electron transfers located on the aromatic system).13 In compound 8, the starting neutral species evolves to the radical-cation and the dication during oxidation and to the radical-anion and dianion during reduction.
 | ||
| Fig. 4 Cyclic voltammetry of compound 7 (bottom) and of fused compound 8 (top): in dichloromethane, 0.1 M NBu4PF6, glassy carbon electrode, 100 mV s−1. | ||
The fused nickel(II) porphyrin 8 (and also the starting compound 7) contains a [6]helicene moiety and consequently presents helical chirality. For both compounds, and also compounds 3 and 4, the enantiomers were separated by chiral HPLC (see ESI† for full experimental conditions). In particular, the two enantiomers of nickel(II) porphyrins 7 and 8 were cleanly separated on a preparative HPLC column ((S,S)-Whelk-O1) and isolated with an ee higher than 98.5%. The electronic circular dichroism (ECD) spectra of the two enantiomers of the two nickel(II) porphyrins 7 and 8 were perfectly symmetrical (Fig. 5). The presence of the extended helicenic aromatic system gave chiroptical properties over the entire UV-visible area of the electronic spectrum.
 | ||
| Fig. 5 Electronic circular dichroism spectra of the two enantiomers of compounds 7 (top) and 8 (bottom). | ||
In summary, the preparation of a fused [6]-helicene-nickel(II) porphyrin molecule was achieved. Due to the thermally stable [6]-helicene moiety, these fused or non-fused molecules presented intrinsic chirality and the separation of the two enantiomers was possible by preparative chiral HPLC. The optical, electrochemical, and chiroptical properties of the fused system were greatly modified when compared to the simply linked helicene-porphyrin molecules. This new chiral chromophore absorbs light over the entire UV-visible region. One important finding for the fusion of helicenes to an aromatic core of a porphyrin was the fact that steric hindrance, due to the approach of the helicene moiety during the cyclodehydrogenation governs the feasibility of the reaction. The cyclisation to form a five-membered ring did not occur with a [6]-helicene substituent, whereas the reaction occurred to form a six-membered ring connection. Work is now in progress to prepare other porphyrinoids fused to one or several helicenes to obtain even larger chiral aromatic chromophores.
Continuous financial support from the CNRS and the University of Strasbourg is acknowledged. VS thanks the French Ministry of Research for his PhD fellowship.
Conflicts of interest
There are no conflicts to declare.Notes and references
- A. Narita, X. Wang, X. Feng and K. Müllen, Chem. Soc. Rev., 2015, 44, 6616–6643 RSC; A. Borissov, Y. K. Maurya, L. Moshniaha, W.-S. Wong, M. Zyla-Karkowska and M. Stepien, Chem. Rev., 2022, 122, 656–788 CrossRef CAS PubMed.
- M. Randic, Chem. Rev., 2003, 103, 3449–3605 CrossRef CAS PubMed.
- M. Rickhaus, M. Mayor and M. Juricek, Chem. Soc. Rev., 2017, 46, 1643–1660 RSC; J. M. Fernandez-Garcia, P. J. Evans, S. Filippone, M. A. Herranz and N. Martin, Acc. Chem. Res., 2019, 52, 1565–1574 CrossRef CAS PubMed; M. A. Majewski and M. Stepien, Angew. Chem., Int. Ed., 2019, 58, 86–116 CrossRef PubMed; M. Krzeszewski, H. Ito and K. Itami, J. Am. Chem. Soc., 2022, 144, 862–871 CrossRef PubMed.
- Z. Qiu, C.-W. Ju, L. Frédéric, Y. Hu, D. Schollmeyer, G. Pieters, K. Müllen and A. Narita, J. Am. Chem. Soc., 2021, 143, 4661–4667 CrossRef CAS PubMed; S. Ma, J. Gu, C. Lin, Z. Luo, Y. Zhu and J. Wang, J. Am. Chem. Soc., 2020, 142, 16887–16893 CrossRef PubMed; P. J. Evans, J. Ouyang, L. Favereau, J. Crassous, I. Fernandez, J. Perles and N. Martin, Angew. Chem., Int. Ed., 2018, 57, 6774–6779 CrossRef.
- Y. Shen and C. F. Chen, Chem. Rev., 2012, 112, 1463–1535 CrossRef CAS PubMed; M. Gingras, Chem. Soc. Rev., 2013, 42, 1051–1095 RSC; C.-F. Chen and Y. Shen, Helicene Chemistry, From Synthesis to Applications, Springer; Berlin, 2017 CrossRef PubMed; E. S. Gauthier, R. Rodriguez and J. Crassous, Angew. Chem., Int. Ed., 2020, 59, 22840–22856 CrossRef PubMed; K. Dhbaibi, L. Favereau and J. Crassous, Chem. Rev., 2019, 119, 8846–8953 CrossRef PubMed.
- T. Hartung, R. Machleid, M. Simon, C. Golz and M. Alcarazo, Angew. Chem., Int. Ed., 2020, 59, 5660–5664 CrossRef CAS PubMed.
- See for example: J. M. Fox, T. J. Katz, S. Van Elshocht, T. Verbiest, M. Kauranen, A. Persoons, T. Thongpanchang, T. Krauss and L. Brus, J. Am. Chem. Soc., 1999, 121, 3453–3459 CrossRef CAS; K. Kato, K. Furukawa, T. Mori and A. Osuka, Chem. – Eur. J., 2018, 24, 572–575 CrossRef PubMed; B. Szyszko, M. Przewoznik, M. J. Bialek, A. Bialonska, P. J. Chmielevski, J. Cichos and L. Latos-Grazynski, Angew. Chem., Int. Ed., 2018, 57, 4030–4034 CrossRef; S. Shimizu, N. Aratani and A. Osuka, Chem. – Eur. J., 2006, 12, 4909–4918 CrossRef; B. Szyszko, P. J. Chmielevski, M. Przewoznik, M. J. Bialek, K. Kupietz, A. Bialonska and L. Latos-Grazynski, J. Am. Chem. Soc., 2019, 141, 6060–6072 CrossRef PubMed; D. C. G. Götz, A. C. Gerhold, S. J. Dorazio, P. Daddario, L. Samankumara, G. Bringmann, C. Brückner and T. Bruhn, Eur. J. Org. Chem., 2015, 3913–3922 CrossRef; C. Brückner, D. C. G. Götz, S. P. Fox, C. Ryppa, J. R. McCarthy, T. Bruhn, J. Akhigbe, S. Banerjee, P. Daddario, H. W. Daniell, M. Zeller, R. W. Boyle and G. Bringmann, J. Am. Chem. Soc., 2011, 133, 8740–8752 CrossRef PubMed; I. Nishimura, T. Higashino and H. Imahori, Chem. Commun., 2021, 57, 9606–9609 RSC; K. Dhbaibi, P. Matozzo, L. Abella, M. Jean, N. Vanthuyne, J. Autschbach, L. Favereau and J. Crassous, Chem. Commun., 2021, 57, 10743–10746 RSC.
- J. P. Lewtak and D. T. Gryko, Chem. Commun., 2012, 48, 10069–10086 RSC; A. M. V. M. Pereira, S. Richeter, C. Jeandon, J.-P. Gisselbrecht, J. Wytko and R. Ruppert, J. Porphyrins Phthalocyanines, 2012, 16, 464–478 CrossRef CAS; T. Tanaka and A. Osuka, Chem. Soc. Rev., 2015, 44, 943–969 RSC.
- A. Tsuda, H. Furuta and A. Osuka, J. Am. Chem. Soc., 2001, 123, 10304–10321 CrossRef CAS PubMed; H. S. Gill, M. Harmjanz, J. Santamaria, I. Finger and M. J. Scott, Angew. Chem., Int. Ed., 2004, 43, 485–490 CrossRef PubMed; M. Tanaka, S. Hayashi, S. Eu, T. Umeyama, Y. Matano and H. Imahori, Chem. Commun., 2007, 2069–2071 RSC; N. K. S. Davis, A. L. Thompson and H. L. Anderson, J. Am. Chem. Soc., 2011, 133, 30–31 CrossRef PubMed; J. P. Lewtak and D. Gryko, Chem. Commun., 2012, 48, 10069–10086 RSC; D. Bao, E. Sebai, O. Vakuliuk, M. Scigaj and D. T. Gryko, Org. Biomol. Chem., 2011, 9, 8178–8181 RSC; J. P. Lewtak, D. Gryko, M. M. Martin, C. Oleszak, F. Hampel and N. Jux, Eur. J. Org. Chem., 2020, 6758–6762 Search PubMed; M. Grzybowski, B. Sadowski, H. Butenschön and D. T. Gryko, Angew. Chem., Int. Ed., 2020, 59, 2998–3027 CrossRef PubMed.
- M. Jakubec, T. Beranek, P. Jakubik, J. Sykora, J. Zadny, V. Cirkva and J. Storch, J. Org. Chem., 2018, 83, 3607–3616 CrossRef CAS; F. B. Mallory and C. W. Mallory, Photocyclizations of stilbenes and related molecules, Organic Reactions, J. Wiley & Sons, Inc., 1984, vol 30, p. 3 Search PubMed.
- Y. Avlasevich, C. Kohl and K. Müllen, J. Mater. Chem., 2006, 16, 1053–1057 RSC; M. Grzybowski, K. Skonieczny, H. Butenschön and D. T. Gryko, Angew. Chem., Int. Ed., 2013, 52, 9900–9930 CrossRef CAS.
- K. Ota, T. Tanaka and A. Osuka, Org. Lett., 2014, 16, 2974–2977 CrossRef CAS PubMed.
- K. M. Kadish, G. Royal, E. Van Caemelbecke and L. Gueletti, in The Porphyrin Hanbook, ed. K. M. Kadish, K. M. Smith, R. Guilard, Academic Press, 2000, vol. 9, pp. 1–219 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Synthetic procedures and characterisation data for new compounds. CCDC 2092869, 2157002, 2157003 and 2157005. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2cc01475k |
| This journal is © The Royal Society of Chemistry 2022 |