
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Visible-light-induced phosgenation of amines by chloroform oxygenation using chlorine dioxide†
Haruyasu
Asahara
 *ab,
Nozomi
Takao
a,
Maiko
Moriguchi
a,
Tsuyoshi
Inoue
ab and
Kei
Ohkubo
*bc
*ab,
Nozomi
Takao
a,
Maiko
Moriguchi
a,
Tsuyoshi
Inoue
ab and
Kei
Ohkubo
*bc
aGraduate School of Pharmaceutical Sciences, Osaka University, 1-6 Yamadaoka, Suita, Osaka 565-0871, Japan. E-mail: asahara@phs.osaka-u.ac.jp
bInstitute for Open and Transdisciplinary Research Initiatives, Osaka University, 1-6 Yamadaoka, Suita, Osaka 565-0871, Japan
cInstitute for Advanced Co-Creation Studies, Osaka University, 1-6 Yamadaoka, Suita, Osaka 565-0871, Japan
First published on 21st April 2022
Abstract
We report the visible-light-induced in situ preparation of COCl2 through the oxygenation of chloroform in the presence of chlorine dioxide, which leads to the safe constructions of carbamoyl chlorides with good-to-high yields and wide substrate scopes. In addition, this method can also be applied to the synthesis of various carbonates.
C1 chemistry is a field of industrial organic chemistry that applies one-C compounds such as CO, CO2, CH4, and CH3OH as the raw materials for transformation reactions, which involve the interconversions of C1 compounds and/or C–C bond formation reactions.1 Among the one-C compounds, halogenated compounds play essential roles in C1 chemistry because of their high reactivities.
The phosgenation reaction, which is one of the most essential organic processes, is widely employed for fine chemical synthesis as well as resin production.2 Among the products obtained from the phosgenation reactions of hetero-nucleophiles, carbamoyl chloride is an essential building block that serves as a precursor for pharmaceutical and agrochemical compounds.3 The application of COCl2,4 a simple and traditional phosgenation reagent and reactive C1 compound, is avoided for the synthesis of these fine chemicals because of the restrictions placed on its application due to its high toxicity. Thus, triphosgene is commonly used as an alternative reagent.5 Triphosgene exists in a stable crystalline form that is safer and easier to transport, store, and handle than COCl2 gas. However, in recent years triphosgene itself has been reported to be highly toxic,6 and an alternative method is urgently needed. The on-demand synthesis of COCl2 through the UV-light irradiation of chloroform (CHCl3) was recently reported,7 which is a simple method that incorporates safe and inexpensive CHCl3 as the solvent and COCl2 precursor. This method requires high-energy UV light, which induces the decomposition of COCl2 as the product as well as the versatility of the substrate. Although reactions with nucleophiles such as alcohols proceed efficiently, they are not suitable for the synthesis of carbamoyl chlorides from light-unstable amines.
On the other hand, we reported the C–H oxygenation reaction of methane (CH4) through the light activation of chlorine dioxide radical (ClO2˙).8 In these oxidation reactions, the chlorine radical (Cl˙) generated from the ClO2˙ gas upon light activation cleaved the C–H bond. The C–H bond dissociation energy of CH4 is 104 kcal mol−1, which was higher than that of CHCl3 (95.7 kcal mol−1).9 These results prompted us to investigate the generation of COCl2 through the oxygenation of CHCl3 with ClO2˙ under visible-light irradiation, because ClO2˙ has a strong absorption band in the visible-light region. Herein, we report the synthesis of carbamoyl chlorides with wide substrate scopes via phosgenation reactions using visible-light irradiation (λ > 400 nm), without decomposing COCl2 (Scheme 1).
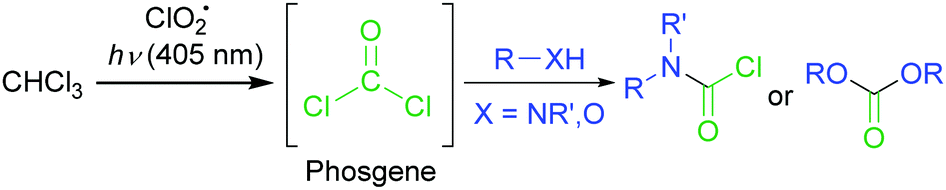 | ||
| Scheme 1 In situ COCl2 preparation through CHCl3 oxygenation using visible-light activated ClO2˙, and further reactions with hetero-nucleophiles to form carbamoyl chloride or carbonate products. | ||
As shown in Fig. 1, an H-shaped reaction glass tube (COware) was employed as the two-chamber system.10 One side of the system (Chamber A, 5 mL) contained an aqueous ClO2˙ solution prepared through the mixing of NaClO2 with HCl. The other side of the system (Chamber B, 2 mL) contained a CHCl3 solution with the substrate. When visible-light irradiation from an LED light (λ = 405 nm) was applied to the whole vessel, gaseous ClO2˙ was generated from Chamber A. The generated ClO2˙ gas transferred through the glass-tube bridge connecting the two chambers, to dissolve in the CHCl3 solution in Chamber B.
 | ||
| Fig. 1 Illustration of the in situ phosgenation process using the two-chamber system. | ||
After 40 min of visible-light irradiation, when the CDCl3 was used without substrate, COCl2 was generated in the CDCl3 and confirmed through an observation of the characteristic signal of its carbonyl carbon at 143 ppm in its 13C NMR spectrum (Fig. S3 in the ESI†).
Encouraged by this result, we commenced the study by employing N-methylaniline 1a as the model amine substrate for reaction condition optimization (Table 1). The target carbamoyl chloride 2a was obtained with an 84% NMR yield when 4 equivalents11 of ClO2˙ and 5 equivalents of NEt3 as the base were applied under the visible-light irradiation of 90 mW cm−2 LED at room temperature (entry 1). A decrease in the visible-light intensity of the reaction decreased the 2a yield to 78% (entry 2), and the reaction did not occur under the dark condition (entry 3). An increase in ClO2˙ effectively afforded 2a with a 93% yield (entry 4). On the other hand, the yield of 2a decreased slightly and a small amount of the urea 3a byproduct was obtained when the ClO2˙ decreased (entry 5). A brief base screening revealed that diisopropylethylamine (DIPEA) was optimal, and the applications of less than two equivalents of DIPEA resulted in poor yields (entries 7–10). Furthermore, we investigated the optimal conditions to obtain urea 3a, and the best results were obtained when ClO2˙ was decreased to one equivalent and pyridine was used as the base (entry 13). It is considered that the pyridine activates the carbamoyl chloride and promotes the addition of a second amine. Because a change in the amount of ClO2˙ or a decrease in the amount of pyridine led to a decrease in the 3a yield, entry 13 was chosen as the optimal reaction condition for urea production.
|
|
|||||||
|---|---|---|---|---|---|---|---|
| Entry | Light (mW cm−2) | Time (min) | ClO2˙ (equiv.) | Base (equiv.) | NMR Yield (%) | ||
| 2a | 3a | ||||||
| a Reaction conditions: 1a (0.2 mmol, 0.1 M), room temperature. | |||||||
| 1 | 90 | 40 | 4 | NEt3 | (5) | 84 | 0 |
| 2 | 30 | 90 | 4 | NEt3 | (5) | 78 | 0 |
| 3 | Dark | 900 | 4 | NEt3 | (5) | 0 | 0 |
| 4 | 90 | 60 | 8 | NEt3 | (5) | 93 | 0 |
| 5 | 90 | 30 | 2 | NEt3 | (5) | 74 | 4 |
| 6 | 90 | 60 | 8 | Pyridine | (5) | 61 | 0 |
| 7 | 90 | 60 | 8 | DIPEA | (5) | 99 | 0 |
| 8 | 90 | 60 | 8 | DIPEA | (3) | 95 | 0 |
| 9 | 90 | 60 | 8 | DIPEA | (2) | 63 | 0 |
| 10 | 90 | 40 | 4 | DIPEA | (3) | 83 | 0 |
| 11 | 90 | 60 | 8 | — | 42 | 0 | |
| 12 | 90 | 30 | 1 | DIPEA | (5) | 16 | 31 |
| 13 | 90 | 30 | 1 | Pyridine | (5) | 0 | 67 |
| 14 | 90 | 40 | 2 | Pyridine | (5) | 0 | 47 |
| 15 | 90 | 20 | 0.5 | Pyridine | (5) | 3 | 39 |
| 16 | 90 | 30 | 1 | Pyridine | (3) | 0 | 34 |

Using the optimized reaction conditions, we investigated the substrate scopes of the phosgenation reactions of N-nucleophiles (Fig. 2). First, the scopes of different aromatic amine (aniline) derivatives were examined. Both anilines with electron-withdrawing and electron-donating substituents afforded their corresponding carbamoyl chlorides (2b and 2c) in good yields. Interestingly, allyl-substituted aniline 1d and iminostilbene 1e underwent phosgenation reactions to afford their desired products in moderate yields and without side reactions such as chlorination of the double alkenyl C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond. However, trace amounts of the product were detected when the diphenylamine 1f was used as the substrate. This is partly owing to the lower nucleophilicity of the 1f compared with those of the N-methyl anilines 1a–c.12 In the case of the conformationally-restricted cyclic derivatives, an unknown byproduct was observed and was likely because of its higher reactivity. Hence, the desired products 2g and 2h were obtained in high yields through a decrease of ClO2˙ to 4 equivalents. We also obtained 2g in good yields when 365 nm LED or sunlight as light source, respectively. Aliphatic amines were compatible in the reactions and afforded the related products in moderate-to-good yields. The phosgenation reactions of dibutyl amine 1i and the cyclic amines 1j and 1k achieved 99, 83, and 58% yields, respectively.13 The proline derivative 1l also afforded the desired product in a moderate yield. The benzyl-substituted amine 1m and the tetrahydroisoquinoline derivatives 1n and 1o were well tolerated under the reaction conditions, and provided the desired products in excellent yields. Notably, the 2o product formed through this method is a key precursor of solifenacin, a competitive cholinergic receptor antagonist. In addition, we tested this method during the late-stage phosgenation reactions of structurally complex pharmaceutical samples. Both of the fluoroquinolone antibiotics, norfloxacin and gatifloxacin, afforded the desired products in high yields and without any detectable side products. When the substrates with nitrogen and oxygen nucleophiles in the same molecule were used, the corresponding cyclic products with inserted carbonyl groups 4–6 were obtained in high yields. The heterocyclic skeletons obtained have been investigated extensively for the developments of various pharmaceuticals and pesticides.14 On the other hand, reactions were complicated when primary amine (toluidine) was used. The expected products, isocyanate or urea, could not be obtained in this reaction conditions.
C bond. However, trace amounts of the product were detected when the diphenylamine 1f was used as the substrate. This is partly owing to the lower nucleophilicity of the 1f compared with those of the N-methyl anilines 1a–c.12 In the case of the conformationally-restricted cyclic derivatives, an unknown byproduct was observed and was likely because of its higher reactivity. Hence, the desired products 2g and 2h were obtained in high yields through a decrease of ClO2˙ to 4 equivalents. We also obtained 2g in good yields when 365 nm LED or sunlight as light source, respectively. Aliphatic amines were compatible in the reactions and afforded the related products in moderate-to-good yields. The phosgenation reactions of dibutyl amine 1i and the cyclic amines 1j and 1k achieved 99, 83, and 58% yields, respectively.13 The proline derivative 1l also afforded the desired product in a moderate yield. The benzyl-substituted amine 1m and the tetrahydroisoquinoline derivatives 1n and 1o were well tolerated under the reaction conditions, and provided the desired products in excellent yields. Notably, the 2o product formed through this method is a key precursor of solifenacin, a competitive cholinergic receptor antagonist. In addition, we tested this method during the late-stage phosgenation reactions of structurally complex pharmaceutical samples. Both of the fluoroquinolone antibiotics, norfloxacin and gatifloxacin, afforded the desired products in high yields and without any detectable side products. When the substrates with nitrogen and oxygen nucleophiles in the same molecule were used, the corresponding cyclic products with inserted carbonyl groups 4–6 were obtained in high yields. The heterocyclic skeletons obtained have been investigated extensively for the developments of various pharmaceuticals and pesticides.14 On the other hand, reactions were complicated when primary amine (toluidine) was used. The expected products, isocyanate or urea, could not be obtained in this reaction conditions.
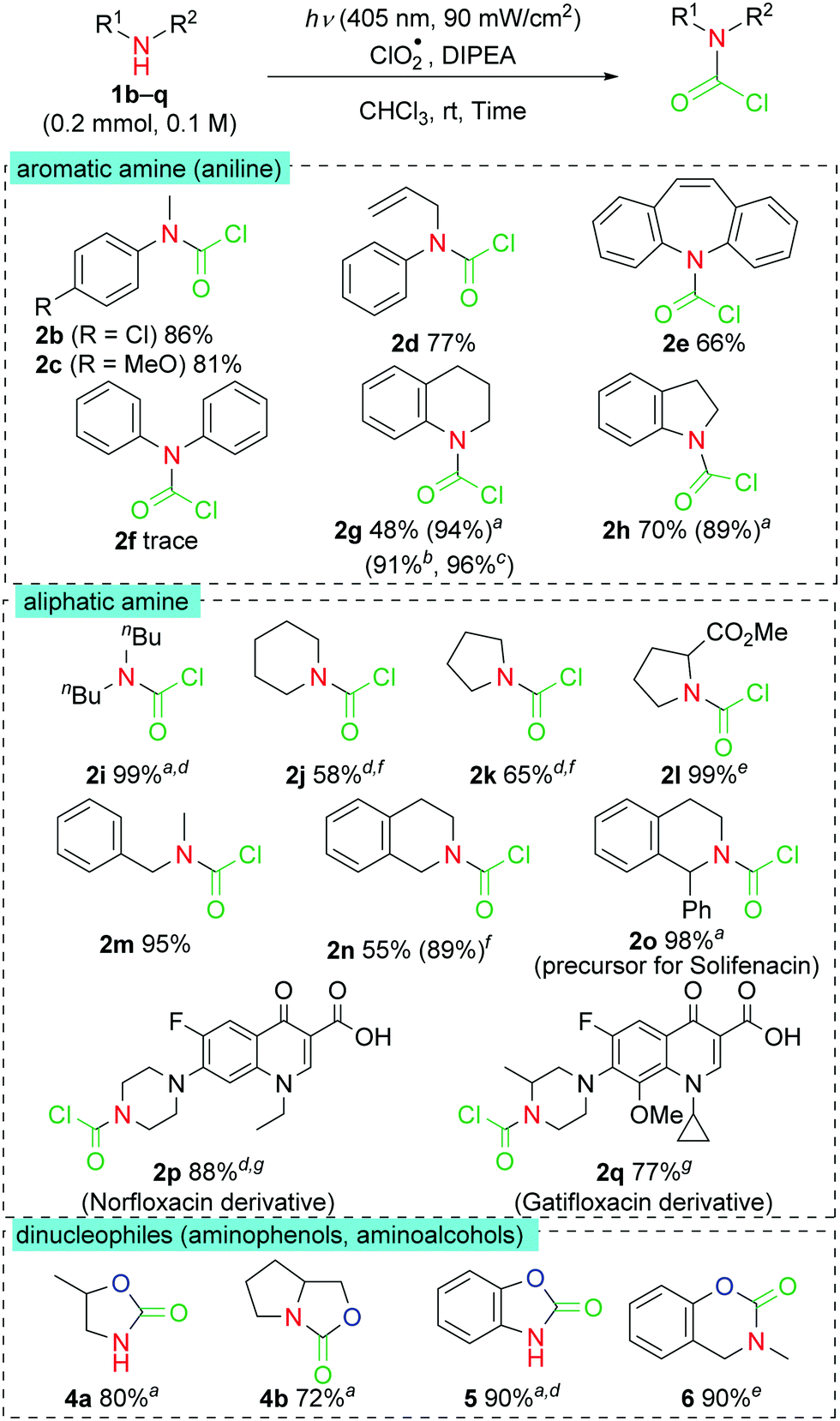 | ||
| Fig. 2 Substrate scopes of the phosgenation reactions of N-nucleophiles. The reactions conditions: amine 1 0.2 mmol (0.1 M), ClO2˙ (8 equiv.), DIPEA (3 equiv.) at room temperature for 60 min. Isolated yields. a ClO2˙ (4 equiv.), 40 min. b 365 nm, ClO2˙ (4 equiv.), 20 min. c Sunlight, ClO2˙ (4 equiv.), 2 h. d Reactions conducted in CDCl3 instead of CHCl3. 1H NMR yields obtained based on the internal standard of 1,1,2,2-tetrachloroethane. e ClO2˙ (4 equiv.), DIPEA (5 equiv.), 40 min. f Amine 1 0.1 mmol (0.02 M), 40 min. g Amine 1 0.1 mmol (0.05 M), ClO2˙ (4 equiv.), 30 min. | ||
We also explored the scopes of these reactions by replacing the nitrogen nucleophiles with oxygen nucleophiles (phenols and alcohols, Fig. 3). The process for the N-methyl aniline was applied to the phenols, and for all their cases, the carbonates 8a–d were obtained in quantitative yields. In addition, the desired carbonates were obtained using the fluorine substituted alcohols as the substrates, although their yields were slightly lower. In the case of n-propanol, carbonate 8g was obtained in moderate yield. It is interesting to note that we also successfully obtained chloroformate 8g′ when 2,6-lutidine was used as a base. This is an important result, although further studies are needed. Diols such as the ethylene glycol, propylene glycol, and catechol derivatives also afforded their cyclic carbonates (9a,b, and 10) at high yields. Different types of carbonates, including diaryl, dialkyl, and cyclic carbonates, are essential in industry and are employed in a broad range of applications15 such as their employments as the starting materials for resin (polycarbonates and polyurethanes) manufacturing, and have recently attracted considerable attention as sustainable process feedstocks.16
 | ||
| Fig. 3 Substrate scopes of the phosgenation reactions of O-nucleophiles. The reactions conditions: phenols or alcohols 7 0.2 mmol (0.1 M), ClO2˙ (4 equiv.), DIPEA (5 equiv.) at room temperature for 40 min.a Reactions conducted in CDCl3 instead of CHCl3. 1H NMR yields obtained based on the internal standard of 1,1,2,2-tetrachloroethane. b Alcohol 7 0.4 mmol (0.4 M), ClO2˙ (2 equiv.), pyridine (5 equiv.). c Alcohol 7 0.1 mmol (0.02 M), ClO2˙ (8 equiv.), 2,6-lutidine (5 equiv.). | ||
To investigate the reaction mechanism of the generation of COCl2 from chloroform by our method, we conducted a control experiment (Scheme S1(a), ESI†). Ethylene glycol, which reacts with COCl2 at a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio, was employed as the substrate and reacted with ClO2˙ (0.5 equiv.) to afford a cyclic carbonate with a 61% yield. This result indicates that an equivalent amount of ClO2˙ is not required for COCl2 formation.
:1 ratio, was employed as the substrate and reacted with ClO2˙ (0.5 equiv.) to afford a cyclic carbonate with a 61% yield. This result indicates that an equivalent amount of ClO2˙ is not required for COCl2 formation.
Furthermore, the product yields of carbonate 9a were determined (Fig. S4, ESI†) with respect to the reaction times in CHCl3 and CDCl3 under the same reaction conditions. It is worth noting that a significant induction period was observed when the reaction was conducted in CDCl3. It has been reported that the difference in bond energies between Cl3C–H and Cl3C–D is 6.0 kcal mol−1.17 These results indicate that hydrogen abstraction from Cl3CH is the rate-limiting step in this reaction.
Based on the experimental results obtained, the DFT calculations performed (M06-2x/6-311 + +G(d,p) level of theory; (see ESI† for detailed protocol), and previous reports,17 a plausible reaction mechanism is presented in Scheme 2. The visible-light activation of ClO2˙ yields chlorine radicals (Cl˙) and singlet oxygen molecules (1O2*) through bond rearrangements from Cl–O–Cl to Cl–O–O bonds.8 The generated Cl˙ abstracts hydrogen from Cl3C–H to form a trichloromethyl radical (Cl3C˙) and HCl.18 This process proceeds more easily than methane oxidation, as indicated by the C–H bond energies (H3C–H: 104 kcal mol−1, Cl3C–H: 95.7 kcal mol−1).9 In fact, the energy difference (ΔE) for this process estimated from DFT calculations is negative (−3.3 kcal mol−1). However, this reaction step is energetically unfavourable for CDCl3 compared with CHCl3, because of its relatively higher bond energy. This could have resulted in a remarkable induction period. The radical intermediate Cl3C˙ then combines with oxygen to produce the peroxyl radical Cl3COO˙.19 The calculated ΔE values for the formations of CCl3OO˙ from the singlet and triplet O2 are −60.5 and −23.1 kcal mol−1, respectively. Hence, once Cl3C˙ is formed, it can rapidly react with both the singlet and triplet O2 to produce the peroxyl radical. Cl3COO˙ gives an alkoxy radical (Cl3CO˙) through the desorption of O2via a Russel-type mechanism, and the ΔE value for this process is estimated to be −4.9 kcal mol−1. There are two possible reaction pathways for the Cl3CO˙; the first pathway is COCl2 formation through the regeneration of Cl˙, and the second pathway is the mechanism of hydrogen abstraction from CHCl3 to form Cl3C˙. Both pathways are estimated to be exothermic with ΔE values of −16.1 and −14.8 kcal mol−1, respectively. The regenerated Cl˙ and Cl3C˙ are recycled to produce COCl2 until the radical chain is terminated. In addition, the generated CCl3OH yields COCl2 along with HCl, and the ΔE value for this step is also negative (−4.5 kcal mol−1). Thus, all the steps after the photochemical generation of Cl˙, as shown in Scheme 2, are exothermic in nature and energetically favourable as a radical chain reaction.20
 | ||
| Scheme 2 Plausible radical chain mechanism for the generation of phosgene. The blue numbers indicate the ΔE values (kcal mol−1) estimated using DFT calculations. | ||
The stoichiometric equation for this oxygenation reaction is given by eqn (1). Two CHCl3 molecules react with one O2 molecule to produce two COCl2 molecules. This means that the ClO2˙ acts as an initiator in the radical chain cycle and as an O2 source for COCl2 formation.
| 2CHCl3 + O2 → 2COCl2 + 2HCl | (1) |
We have developed a visible-light-induced COCl2 generation method using CHCl3 and sodium chlorite as the starting materials, which are inexpensive and easy to handle. Various carbamoyl chlorides can be synthesized safely and efficiently via the phosgenation reactions of amines using COCl2 generated in situ. This is an excellent method that can be applied to a wide range of substrates, including anilines and aliphatic amines, as well as pharmaceutical compounds with nucleophilic nitrogen atoms. In addition, this phosgenation method was successfully applied to carbonate synthesis from phenols and alcohols. This novel phosgenation system is an alternative to the classical method that involves the use of a hazardous reagent.
This work was supported by JSPS KAKENHI (JP20K05606 to H. A., JP20H02779 and JP20H04819 to K. O.); the NEDO 17101509-0 to H. A.; and the JST OPERA JPMJOP1861 to T. I.
Conflicts of interest
The authors declare no conflict of interest.Notes and references
- (a) C. Mesters, Annu. Rev. Chem. Biomol. Eng., 2016, 7, 223 CrossRef PubMed; (b) W. Zhou, K. Cheng, J. Kang, C. Zhou, V. Subramanian, Q. Zhang and Y. Wang, Chem. Soc. Rev., 2019, 48, 3193 RSC; (c) Y. Liu, D. Deng and X. Bao, Chemistry, 2020, 6, 2497 CrossRef CAS.
- (a) H. Babad and A. G. Zeiler, Chem. Rev., 1973, 73, 75 CrossRef CAS; (b) L. Cotarca and H. Eckert, Phosgenations-A Handbook, Wiley-VCH; Weinheim, 2003 CrossRef.
- (a) D. R. Lide, CRC Handbook of Chemistry and Physics, CRC Press;Boca Raton, FL, 87th edn, 2007, pp. 4002 Search PubMed; (b) P. Jaeger, C. N. Rentzea and H. Kieczka, Ullmann's encyclopedia of industrial chemistry, Wiley-VCH; Weinheim, Germany, 2012 Search PubMed; (c) K. Adeppa, D. C. Rupainwar and K. Misra, Can. J. Chem., 2010, 88, 1277 CrossRef CAS; (d) P. Jaeger, C. N. Rentzea and H. Kieczka, Ullmann's encyclopedia of industrial chemistry, Wiley-VCH; Weinheim, Germany, 2014, pp. 399 Search PubMed; (e) M. Shrestha, X. Wu, W. Huang, J. Qu and Y. Chen, Org. Chem. Front., 2021, 8, 4024–4045 RSC.
- Global Phosgene Outlook 2016-2021, Gen Consulting Company, 2016.
- (a) H. Eckert and B. Förster, Angew. Chem., Int. Ed. Engl., 1987, 26, 894 CrossRef; (b) L. Cotarca, P. Delogu, A. Nardelli and V. Sunjic, Synthesis, 1996, 553 CrossRef CAS; (c) W. Su, W. Zhong, G. Bian, X. Shi and J. Zhang, Org. Prep. Proced. Int., 2004, 36, 499 CrossRef CAS; (d) M. O. Ganiu, B. Nepal, J. P. Van Houten and R. Kartika, Tetrahedron, 2020, 76, 131553 CrossRef CAS PubMed.
- H. Eckert, Chim. Oggi, 2011, 29, 40 CAS.
- Y. Kuwahara, A. Zhang, H. Soma and A. Tsuda, Org. Lett., 2012, 14, 3376 CrossRef CAS PubMed.
- (a) K. Ohkubo and K. Hirose, Angew. Chem., Int. Ed., 2018, 57, 2126 CrossRef CAS PubMed; (b) K. Ohkubo, H. Asahara and T. Inoue, Chem. Commun., 2019, 55, 4723 RSC.
- J. A. Kerr, Chem. Rev., 1966, 66, 465 CrossRef CAS.
- For examples using two-chamber system, see: (a) X. Jia, S. Kramer, T. Skrydstrup and Z. Lian, Angew. Chem., Int. Ed., 2021, 60, 7353 CrossRef CAS PubMed; (b) L. Chen, M. Zhou, L. Shen, X. He, X. Li and X. Zhang, Org. Lett., 2021, 23, 4991 CrossRef CAS PubMed; (c) K. Nozawa-Kumada, K. Noguchi, T. Akada, M. Shigeno and Y. Kondo, Org. Lett., 2021, 23, 6659 CrossRef CAS PubMed.
- Based on the amount of sodium nitrite (0.8 equivalent of ClO2˙ was generated from ClO2 – by dismutation reaction).
- J. J. Campbell and S. A. Glover, J. Chem. Res., 1999, 474 CAS.
- Reaction was conducted in CDCl3 due to the low boiling point of the substrate. In addition, the concentration of amines 1j,k was reduced to suppress the formation of corresponding urea.
- (a) D. Chaturvedi, Tetrahedron, 2012, 68, 15 CrossRef CAS; (b) K. I. Reddy, C. Aruna, S. K. Babu, V. Vijayakumar, M. Manisha, J. P. Sridevi, P. Yogeeswari and D. Sriram, RSC Adv., 2014, 4, 59594 RSC; (c) K. E. Ryu, B. R. Kim, G. H. Sung, H. J. Yoon and Y.-J. Yoon, Synlett, 2015, 1985 CAS; (d) Y. Chen, C. A. Hone, B. Gutmann and C. O. Kappe, Org. Process Res. Dev., 2017, 21, 1080 CrossRef CAS; (e) J. Lee, J. Lee, H. Jung, D. Kim, J. Park and S. Chang, J. Am. Chem. Soc., 2020, 142, 12324 CrossRef CAS PubMed.
- (a) F. Bigi, R. Maggi and G. Sartori, Green Chem., 2000, 2, 140 RSC; (b) M. Carafa, V. Melea and E. Quaranta, Green Chem., 2012, 14, 217 RSC; (c) S. Huang, B. Yan, S. Wang and X. Ma, Chem. Soc. Rev., 2015, 44, 3079 RSC; (d) E. R. Baral, J. H. Lee and J. G. Kim, J. Org. Chem., 2018, 83, 11768 CrossRef CAS PubMed.
- (a) O. Kreye, H. Mutlu and M. A. R. Meier, Green Chem., 2013, 15, 1431 RSC; (b) Z. Dobi, B. N. Reddy, E. Renders, L. Van Raemdonck, C. Mensch, G. De Smet, C. Chen, C. Bheeter, S. Sergeyev, W. A. Herrebout and B. U. W. Maes, ChemSusChem, 2019, 12, 3103 CrossRef CAS PubMed; (c) M. Soccio, R. Mazzoni, C. Lucarelli, S. Quattrosoldi, A. Cingolani, M. Fiorini, N. Lotti and T. Tabanelli, ACS Sustainable Chem. Eng., 2020, 8, 15640 CrossRef CAS.
- (a) T. Alapi and A. Dombi, Chemosphere, 2007, 67, 693 CrossRef CAS PubMed; (b) A. J. Seidl, L. R. Cohen, L. A. Peña and P. E. Hoggard, Photochem. Photobiol. Sci., 2008, 7, 1373 CrossRef CAS PubMed.
- In experiments using TEMPO as a radical trap reagent, inhibition of the reaction was observed (Scheme. S1(b), ESI†).
- A radical intermediate, Cl3COO˙ was detected under photoirradiation of a chloroform solution containing ClO2˙ by ESR spectroscopy (see Fig. S5 in ESI†).
- When light irradiation was turned off 5 minutes after the reaction started, an increase in product (10 to 35%) was observed even under shielded light (see Fig. S4 in ESI†).
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details and characterization data. See DOI: https://doi.org/10.1039/d2cc01336c |
| This journal is © The Royal Society of Chemistry 2022 |