
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
UV degradation of poly(lactic acid) materials through copolymerisation with a sugar-derived cyclic xanthate†
Craig
Hardy
 a,
Gabriele
Kociok-Köhn
b and
Antoine
Buchard
*a
a,
Gabriele
Kociok-Köhn
b and
Antoine
Buchard
*a
aCentre for Sustainable and Circular Technologies, Department of Chemistry, University of Bath, Bath, BA2 7AY, UK. E-mail: a.buchard@bath.ac.uk
bMaterials and Chemical Characterisation Facility (MC2), University of Bath, UK
First published on 6th April 2022
Abstract
The copolymerisation of L-Lactide with a cyclic xanthate monomer derived from tri-O-acetyl-D-glucal has been used to incorporate thionocarbonate and thioester linkages into a polyester backbone. The poly(lactide-co-xanthate) copolymers show enhanced UV-degradability compared to PLA, with 40% mass loss within 6 hours of UV exposure (365 nm) for only 3% of sulfur-containing linkages.
Amidst the environmental persistence and reliance on fossil-based resources of the most common polymers, polymers based on renewable resources, and which can be degraded, have been investigated as alternatives.1–3 Poly(lactic acid) (PLA) is a thermoplastic aliphatic polyester derived from lactic acid and is perhaps the most widely studied degradable and renewable polymer. While PLA degrades under industrial composting conditions (62 ± 4 °C, >60% relative humidity) within weeks, it lacks facile degradability in natural environments, e.g. in soil or in seawater.4,5 One strategy to adjust the degradation of PLA has been the copolymerisation of lactide (LA) with other monomers resulting in linkages more susceptible to hydrolysis.4,6 Once oligomeric, PLA indeed dissolves quicker and is attacked by enzymes more effectively. Thus, in biomedical applications, poly(lactic-co-glycolic acid) (PLGA) is a common material.7 PLA hydrolytic degradation has also been enhanced recently by copolymerisation with cyclic phosphoester8 and salicylates.9 However, to the best of our knowledge, no modification of PLA has been directed at enhancing its degradability towards stimuli other than hydrolysis, such as light.
As part of our wider research programme on renewable polymers, we, amongst others, have been investigating the incorporation of monosaccharide units into polymer backbones. The natural abundance, structural diversity and functionalisation potential of sugars make them a renewable resource with tremendous potential for sustainable materials.10,11 Of relevance here, we have thus reported the ring-opening copolymerisation of a xylose-based oxetane with CS2, which yield polythiocarbonates that rapidly degrade under UV radiation.12 We also previously reported the Ring-Opening Polymerisation (ROP) of cyclic xanthates from C5 sugars (D-ribose and D-xylose).13 However, their synthesis was challenging, with extensive purification needed for ROP. Herein, our hypothesis has been that the copolymerisation of lactide with a cyclic xanthate would allow the incorporation of UV-sensitive linkages into PLA. We report the synthesis of a novel cyclic xanthate monomer derived from D-glucal, its ring-opening polymerisation (ROP) and copolymerisation with L-lactide, which results in unprecedented UV-degradable poly(lactic acid).
To circumvent the difficulties met previously, we selected tri-O-acetyl-D-glucal as starting material, a commercial derivative of C6 sugar D-glucose, previously used for polycarbonate synthesis by ROP. One attractive feature of glucal-derived monomers is the incorporation in the polymer backbone of a cyclic alkene, primed for post-polymerisation modifications. Building on the strategy developed by Wooley and coworkers,14 the synthesis of the desired cyclic xanthate involved four high-yielding synthetic steps (59% yield) from tri-O-acetyl-D-glucal: Ferrier rearrangement, deprotection under Zemplén conditions, tosylation at the C-6 position, then cyclisation using CS2 (Scheme 1).
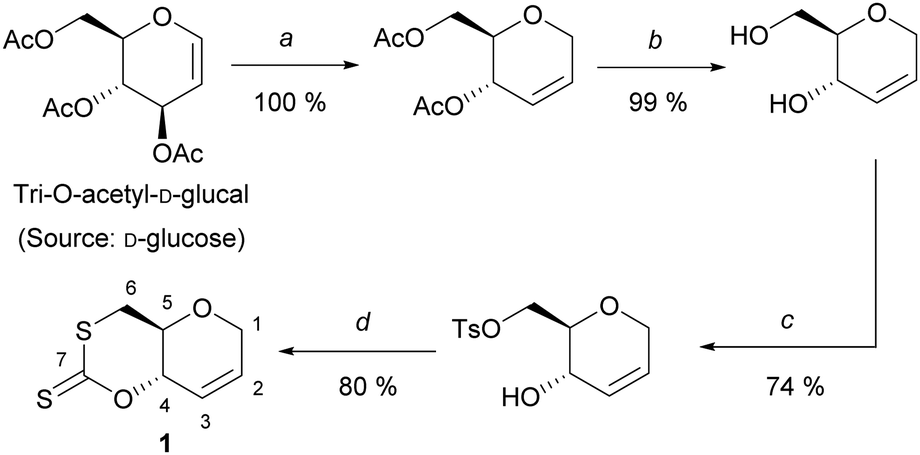 | ||
| Scheme 1 Synthesis of 1; (a) Et3SiH, BF3OEt2, DCM, 0 °C, 3 h; (b) NaOMe, MeOH, rt, 2 h; (c) TsCl, Pyridine, rt, 20 h; (d) CS2, t-BuOK, THF, 0 °C, 3 h. | ||
After recrystallisation in ethanol, cyclic xanthate 1, was isolated as pale-yellow crystals, and was characterized by multinuclear NMR and FT-IR spectroscopies. Unambiguously, 13C{1H} NMR analysis identified a signal at δC ≈ 208 ppm, characteristic of the C(S)SO linkage of cyclic xanthates.15 Molecular structure and stereochemistry were further corroborated by single crystal X-ray diffraction crystallography (Fig. S23, ESI†).
The ROP of 1 was successfully conducted at room temperature (rt) in dichloromethane (DCM), using 1,5,7-triazabicyclo[4.4.0]dec-5-ene (TBD) as catalyst, 4-methylbenzyl alcohol (4-MeBnOH) as initiator (Table 1). Early trials with other organocatalysts indeed resulted in lower activities (ESI† Table S1). At 1 mol L−1 initial monomer concentration, ROP proceeded readily, rapidly reaching a plateau, with only marginal increase in monomer conversion when increasing reaction times from 6 h (71–72%; Table 1, entries 1–3) to 24 h (77–78%; Table 1, entries 4–6). This plateau in monomer conversion is indicative of an equilibrium limited opening of the ring and is consistent with the exceptional stability (towards air, moisture, and heat) of cyclic xanthate 1. This is in stark contrast with the analogous 6-membered cyclic carbonates, for which the ROP equilibrium lies heavily towards the polymeric products (>99%),14,16–18 highlighting how exchanging oxygen for sulfur atoms strongly impacts ring strain and polymerisability.
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | [1]0![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :[TBD]0:[I]0b :[TBD]0:[I]0b |
Time (h) | Conv (%)c | M n,calc | M n,NMR | M n,SEC [ĐM]f |
| a Polymerisations were carried out at rt, under an argon atmosphere, in anhydrous CH2Cl2 solvent with initial [1]0 = 1 mol L−1 (1 = monomer), unless stated otherwise. b I = 4-methylbenzylalcohol. c Monomer conversion to polymer, calculated based on the relative integration of the H-4′ proton signal of 1 (δH = 4.79 ppm) and poly(1) (δH = 5.75–5.68 ppm), in the 1H NMR spectrum. d Number-average molar mass as calculated using Mr(I) + (Mr(monomer) × [monomer]0/[I]0 × conv/100%) (kg mol−1). e Number-average molar mass calculated by the relative integration of the aromatic protons in I (δ = 7.30–7.15 ppm, 4 H) and H-4′ in poly(1) (kg mol−1). f Number-average molar mass and Dispersity (Mn,SEC, ĐM), calculated by SEC relative to polystyrene standards in THF eluent (kg mol−1). g [1]0 = 0.5 mol L−1. h [1]0 = 2 mol L−1. | ||||||
| 1 | 100:1:1 |
6 | 71 | 13.5 | 9.2 | 6.8 [1.73] |
| 2 | 50:1:1 |
6 | 71 | 6.8 | 5.5 | 5.2 [1.58] |
| 3 | 25:1:1 |
6 | 72 | 3.5 | 2.8 | 4.0 [1.44] |
| 4 | 100:1:1 |
24 | 77 | 14.6 | 10.1 | 8.7 [1.75] |
| 5 | 50:1:1 |
24 | 77 | 7.4 | 5.6 | 5.9 [1.69] |
| 6 | 25:1:1 |
24 | 78 | 3.8 | 2.8 | 4.7 [1.51] |
| 7g | 100:1:1 |
24 | 58 | 9.7 | 5.9 | 4.3 [1.58] |
| 8h | 100:1:1 |
24 | 90 | 17.4 | 8.1 | 10.0 [1.68] |
| 9 | 150:1:1 |
24 | 69 | 19.6 | 13.6 | 10.5 [1.68] |
| 10 | 200:1:1 |
24 | 75 | 28.4 | 15.9 | 10.7 [1.76] |
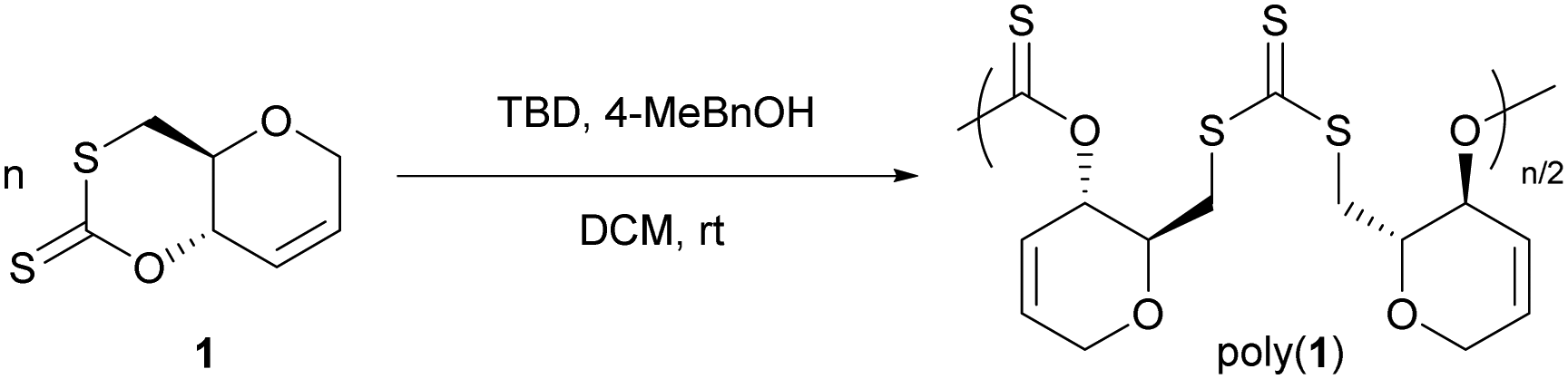
As expected from an equilibrium-limited ROP process, initial monomer concentration could be manipulated to reach higher monomer conversion, and 90% conversion was achieved for [1]0 = 2.0 mol L−1 (Table 1, entry 8). Interestingly for polymer chemical recyclability, a near quantitative recovery of the monomer is observed when poly(1) is stirred in the presence of TBD in dilute conditions (0.1 mol L−1). The temperature dependence of the ROP equilibrium was also investigated in 1,2-dichloroethane (DCE) at fixed initial monomer concentration, catalyst and initiator loadings. Decreasing temperature from 60 °C to 0 °C resulted in an increase in monomer conversion from 2% up to 85%. Below 0 °C 1 was no longer soluble resulting in no reaction. A plot of ln([1]eq) vs. 1/T gave an approximation of the polymerisation thermodynamic parameters (Fig. S46, ESI†): ΔHp = −25.6 kJ mol−1 and ΔSp = −80 J mol−1. A similar study using THF instead of DCE also gave an estimation of these parameters in THF (Fig. S47, ESI†; ΔHp = −22.0 kJ mol−1 and ΔSp = −61 J mol−1 K−1; revealing a moderate increase in the enthalpy of polymerisation, but with little impact on polymerisability.
Upon reaction completion, the polymer products could be isolated by precipitation in diethyl ether, and SEC was utilised to estimate number-average molar masses (Mn) and dispersities (ĐM). Discrepancy between expected molar masses and those calculated by SEC was found (Table 1), in addition to limitations in obtaining high molar mass polymers, which both may be due to impurities in the monomer acting as chain-transfer agents. Nevertheless, polymers of up to 10700 g mol−1 (ĐM 1.76) could be obtained, some control of the polymerisation was indicated by the linear relationship between monomer conversion and Mn (Fig. S45, ESI†) and first-order kinetics with respect to monomer concentration (Fig. S40–S44, ESI†).
13C{1H} NMR spectroscopy was primarily used to investigate the microstructure of poly(1) (Fig. 1). Two major thiocarbonyl resonances at δC ≈ 194 and 223 ppm, of similar intensity, could be seen and were assigned to a mono-thionocarbonate (C(S)O2) environment and a tri-thiocarbonate environment (C(S)S2), respectively.12,13 Only a small amount of xanthate linkages (SC(S)O; 1–2%) could be identified from a minor resonance at δC ≈ 214 ppm.191H NMR spectra also featured a CH–O (δH = 5.68–5.75) and a shielded CH2–S (δH = 3.68–3.58) proton environments (Fig. S24, ESI†). Collectively, this data suggests the ROP of 1 proceeds via the alternating opening of 1 on either side of the xanthate thiocarbonyl, and subsequent selective chain propagation, yielding regioregular polymers with alternating C(S)S2 and C(S)O2 linkages, similar to what we observed for a xylose-based cyclic xanthate.
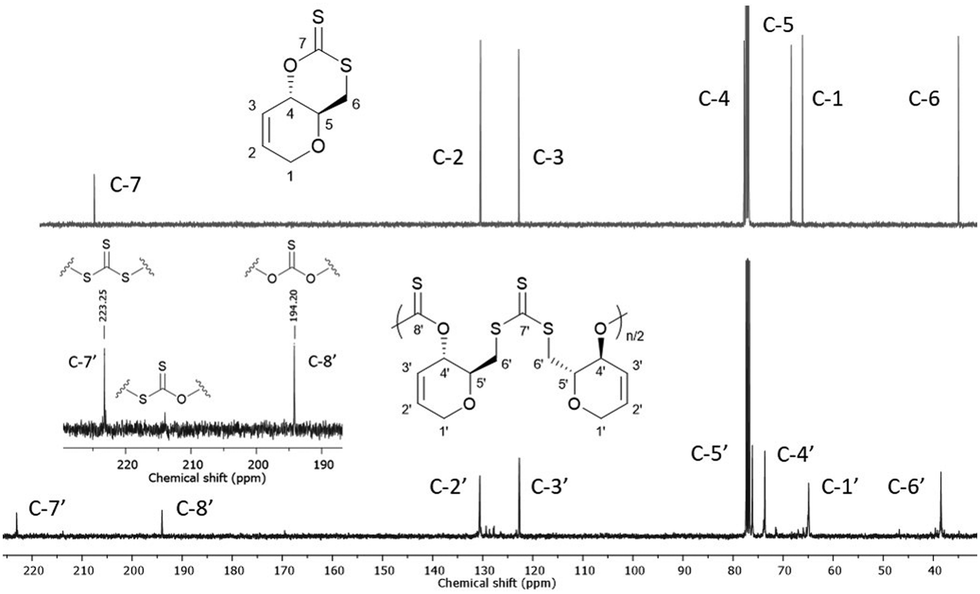 | ||
| Fig. 1 13C{1H} NMR spectra (CDCl3) of 1 (top) and poly(1) (bottom) (Table 1, entry 4). (inset) Alternating C(S)S2 and C(S)O2 linkages observed for poly(1). | ||
While MALDI-ToF mass spectrometry proved unsuccessful to probe further the macromolecular structure of poly(1), a good agreement between Mn,NMR (estimated by end-group analysis) and Mn,SEC values suggested the absence of major cyclic polymer species. When the ROP of 1 was carried out above 50 °C, especially in THF, a vibration at 1709 cm−1 in the FTIR spectra, characteristic of a carbon–oxygen double bond (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) vibrational mode (Fig. S52, ESI†), emerged. Two new carbonyl resonances could also be observed by 13C{1H} NMR spectroscopy, at 169 and 154 ppm, assigned to mono-thiocarbonate (C(O)SO) and carbonate (C(O)O2) environments, respectively (Fig. S48, ESI†). This was indicative of some S–O exchange reactions, which in situ IR spectroscopy showed to occur after maximum monomer conversion is reached.
O) vibrational mode (Fig. S52, ESI†), emerged. Two new carbonyl resonances could also be observed by 13C{1H} NMR spectroscopy, at 169 and 154 ppm, assigned to mono-thiocarbonate (C(O)SO) and carbonate (C(O)O2) environments, respectively (Fig. S48, ESI†). This was indicative of some S–O exchange reactions, which in situ IR spectroscopy showed to occur after maximum monomer conversion is reached.
Poly(1)s were shown to be amorphous by differential scanning calorimetry (DSC) with a glass transition temperature (Tg) of 65 °C (Fig. S33–S36, ESI†; polymers of Mn 10 700 g mol−1 (ĐM 1.8) and 6800 g mol−1 (ĐM 1.6)). Thermogravimetric analysis (TGA) revealed the onset of thermal degradation (Td5%) to occur around 188 °C, with a final residual char of 19 and 15%, respectively (Figs. S37 and S38, ESI†).
Copolymerisation of 1 with L-lactide (L-LA) was first investigated through sequential monomer addition. 25 equivalents of 1 were first polymerised (DCM, rt; [1]0:[TBD]0:[MeBnOH]0 = 25:1:1; [1]0 = 1.0 mol L−1). Then, after 6 hours and 78% conversion of 1 (Mn 4400 g mol−1), 50 equivalents of L-LA were added to the reaction alongside some additional TBD (1.0 equiv.). After 2 hours, 1H NMR spectroscopy showed 81 and 98% conversion of 1 and L-LA, respectively. After isolation of the polymer by precipitation, NMR spectroscopy confirmed the formation of a diblock copolymer (Fig. S57–S62, ESI†), while demonstrating the livingness of the ROP process (poly(1-b-LLA), Mn 17 300 g mol−1; Fig. S63, ESI†).
One-pot copolymerisation of 1 with L-LA was also performed (Table 2). L-LA was quantitatively converted while as before, 1 reached a conversion plateau between 71–78%. 1H and DOSY NMR analysis revealed the formation of a copolymer and its composition, which diverged from the monomers feed ratio as the proportion of 1 increased. In the carbonyl region of the 13C{1H} NMR spectra, alongside a major PLA ester signal at δC ≈ 169 ppm, all copolymers exhibited multiple thiocarbonyl resonances at δC ≈ 194 ppm, consistent with thionocarbonate (C(S)O2) and thioester (C(O)S) environments (Fig. 2). As no xanthate or trithiocarbonate signals were observed, the probability of consecutively opening two or more molecules of 1 was deemed low. We propose that the incorporation of 1 in the copolymer mainly proceeds via the attack of 1 by a growing PLA chain and the cleavage of its C–S bond, producing a primary thiol which next opens a LA molecule. Copolymers with consecutive thionocarbonate and thioester linkages inserted within a polyester backbone are thus formed (poly(1-co-LLA)).
|
|
|||||||
|---|---|---|---|---|---|---|---|
| Entry | f 1 /fLLA | Conv. 1 (%) b | Conv. LLA (%)c | M n,SEC [ĐM] d | F 1 /FLLAe | T g (°C)f | T d5% (°C) |
|
a Polymerisations were carried out at rt, for 20 h, under an argon atmosphere, in anhydrous CH2Cl2 solvent with initial [Mt]0 = 1 mol L−1 (Mt = 1 + L-LA) and [Mt]0:[TBD]0:[4-MeBnOH]0 = 100:1:1.
b Monomer conversion to polymer, calculated based on the relative integration of the H-4′ proton signal of 1 (δH = 4.79 ppm) and poly(1) (δH = 5.75–5.68 ppm), in the 1H NMR spectrum.
c Monomer conversion to polymer, calculated based on the relative integration of the lactide alkoxy proton signals in the crude 1H NMR spectrum.
d Number-average molecular weight and Dispersity (Mn,SEC, ĐM), calculated by SEC relative to polystyrene standards in THF eluent (kg mol−1).
e Copolymer compositions determined by integration of the 1H NMR spectra of the purified copolymers.
f Values taken from DSC second heating cycle.
|
|||||||
| 1 | 2/98 | 71 | 98 | 22.5 [1.59] | 1/99 | 51 | 228 |
| 2 | 5/95 | 71 | 99 | 13.2 [1.66] | 3/97 | 51 | 222 |
| 3 | 10/90 | 71 | 99 | 14.8 [1.70] | 6/94 | 49 | 223 |
| 4 | 20/80 | 77 | 98 | 15.8 [1.80] | 16/84 | 47 | 187 |
| 5 | 50/50 | 71 | 98 | 25.1 [1.86] | 29/71 | 50 | 200 |
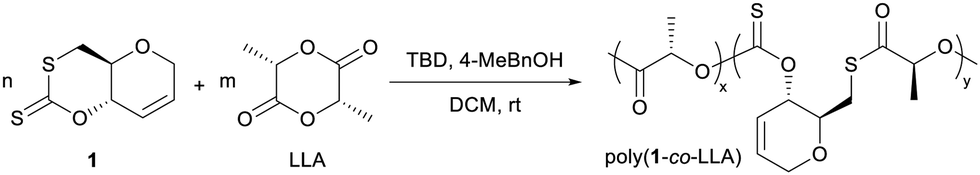
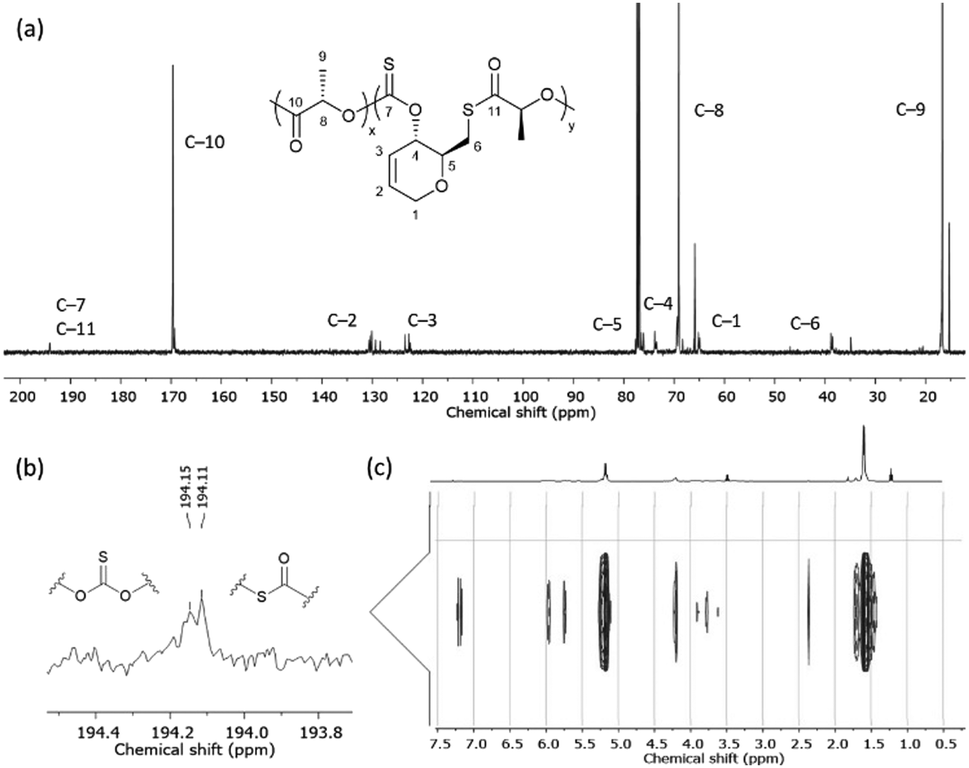 | ||
| Fig. 2 (a) 13C{1H} NMR spectrum (CDCl3) with (b) C(S)O2 and C(O)S linkages observed for poly(1-co-LLA), and (c) 1H DOSY NMR spectrum (CDCl3) of isolated poly(1-co-LLA) (Table 2, entry 4). | ||
Even with as little as 1% 1 content, all poly(1-co-LLA)s were shown to be amorphous by DSC analysis (Table 2; Fig. S72–S76, ESI†), with Tgs ranging, with no identifiable trend, from 47 to 51 °C. TGA also revealed no obvious trend between the compositions of poly(1-co-LLA)s and their thermal stability, with Td5% between 187 and 228 °C (Table 2; Fig. S77–S81, ESI†).
Building upon previous studies surrounding photodegradation,20,21 as well as our experience with similar poly(thiocarbonates), poly(1) could be readily degraded under UV radiation (λ = 365 nm) in THF, with or without tris-(trimethylsilyl)silane (TTMSS) to accelerate the reaction (Fig. S85 and S86, ESI†).12,22 Analysis of the degradation products by 1H NMR spectroscopy proved difficult, although the absence of the H-6’ proton signals suggested the complete breakdown of the trithiocarbonate linkages.
The degradation of statistical copolymers poly(1-co-LLA)s under UV irradiation in THF, in the absence of TTMSS, was also investigated (Fig. 3, polymers from entries 2 and 4, Table 2). Satisfyingly, a gradual decrease in molar mass was observed, which correlated well with the quantity of thionocarbonate/thioester linkages in the polymer. Analysis of the degradation products by 1H NMR spectroscopy revealed a breakdown of the thionocarbonate/thioester linkages leaving predominantly PLLA species (Fig. S87 and S88, ESI†). Under identical conditions, no change was observed for PLLA, supporting the hypothesis that degradation is due to sulfur-containing linkages. UV-Vis spectra of the polymers (Fig. S82 and S83, ESI†) displayed a singular absorbance within the ultraviolet region (λmax ≈ 306 nm) confirming that UV irradiation is the stimulus promoting photodegradation. Photodegradation of PLA has been relatively unexplored, and often requires the incorporation of light responsive nanoparticles or the use of a nucleation agent, as well as extended periods of time (days/weeks) to reach significant levels of molar mass reduction (>20%).23–25 Here, over 40% mass loss within 6 hours of UV exposure was seen, including for copolymers with as little as 3% thionocarbonate/thioester linkages.
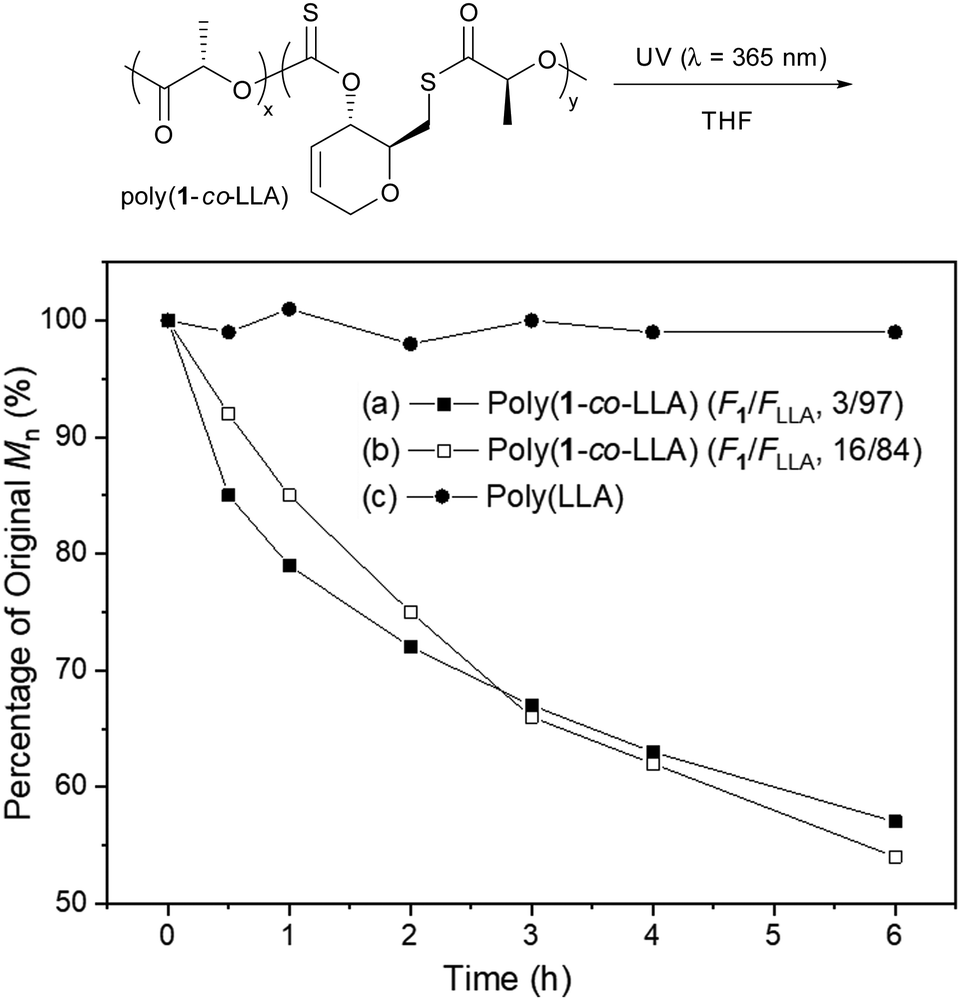 | ||
| Fig. 3 Percentage of original Mnvs. time, following the exposition of poly(1-co-LLA) copolymers to UV irradiation (λ = 365 nm). The reactions were performed at rt with [poly(1-co-LLA)]0 = 0.040 mol L−1. (a) Poly(1-co-LLA) (F1/FLLA, 3/93) (Mn,SEC = 13,200 g mol−1, ĐM = 1.66), (b) poly(1-co-LLA) (F1/FLLA, 16/84) (Mn,SEC = 15,800 g mol−1, ĐM = 1.80) and (c) poly(LLA) (Mn,SEC = 12,800 g mol−1, ĐM = 1.12). | ||
In summary, we have synthesised a novel, bio-derived, unsaturated xanthate cyclic monomer, reported its ring-opening polymerisation and characterised the resulting poly(thiocarbonate)s. The sugar-based monomer was also copolymerised with L-lactide towards block and statistical copolymers. Even at low content, introducing UV-cleavable sulfur-containing linkages into PLA enabled its effective photodegradation into biodegradable oligomeric species. The mechanism of degradation requires to be investigated in detail, and this strategy remains to be translated to real-life plastics objects, but it has the potential to increase the environmental degradability of PLA without significantly affecting its material properties.
We thank the Royal Society (UF\160021, fellowship to AB; RGF\EA\201023; RGF\EA\180028, studentship to CH) for research funding.
Conflicts of interest
There are no conflicts of interest to declare.Notes and references
- R. Geyer, J. R. Jambeck and K. L. Law, Sci. Adv., 2017, 3, e1700782 CrossRef PubMed.
- J.-G. Rosenboom, R. Langer and G. Traverso, Nat. Rev. Mater., 2022, 7, 117–137 CrossRef PubMed.
- S. Walker and R. Rothman, J. Cleaner Prod., 2020, 261, 121158 CrossRef CAS.
- P. McKeown and M. D. Jones, Sustainable Chem., 2020, 1, 1–22 CrossRef.
- S. M. Emadian, T. T. Onay and B. Demirel, Waste Manage., 2017, 59, 526–536 CrossRef CAS PubMed.
- N. F. Zaaba and M. Jaafar, Polym. Eng. Sci., 2020, 60, 2061–2075 CrossRef CAS.
- H. K. Makadia and S. J. Siegel, Polymers, 2011, 3, 1377–1397 CrossRef CAS PubMed.
- T. Rheinberger, J. Wolfs, A. Paneth, H. P.Paneth and F. R. Wurm, J. Am. Chem. Soc., 2021, 143, 15784–15790 CrossRef PubMed.
- H. J. Kim, M. Hillmyer and C. Ellison, J. Am. Chem. Soc., 2021, 143, 15784–15790 CrossRef CAS PubMed.
- G. L. Gregory, E. M. López-Vidal and A. Buchard, Chem. Commun., 2017, 53, 2198–2217 RSC.
- S. L. Kristufek, K. T. Wacker, Y.-Y. T. Tsao, L. Su and K. L. Wooley, Nat. Prod. Rep., 2017, 34, 433–459 RSC.
- T. M. McGuire and A. Buchard, Polym. Chem., 2021, 12, 4253–4261 RSC.
- E. M. López-Vidal, G. L. Gregory, G. Kociok-Köhn and A. Buchard, Polym. Chem., 2018, 9, 1577–1582 RSC.
- A. T. Lonnecker, Y. H. Lim and K. L. Wooley, ACS Macro Lett., 2017, 6, 748–753 CrossRef CAS.
- M. Luo, X.-H. Zhang and D. J. Darensbourg, Macromolecules, 2015, 48, 5526–5532 CrossRef CAS.
- K. Mikami, A. T. Lonnecker, T. P. Gustafson, N. F. Zinnel, P.-J. Pai, D. H. Russell and K. L. Wooley, J. Am. Chem. Soc., 2013, 135, 6826–6829 CrossRef CAS PubMed.
- Y. Song, X. Ji, M. Dong, R. Li, Y.-N. Lin, H. Wang and K. L. Wooley, J. Am. Chem. Soc., 2018, 140, 16053–16057 CrossRef CAS PubMed.
- Y. Song, X. Yang, Y. Shen, M. Dong, Y.-N. Lin, M. B. Hall and K. L. Wooley, J. Am. Chem. Soc., 2020, 142, 16974–16981 CrossRef CAS PubMed.
- D. J. Darensbourg, J. R. Andreatta, M. J. Jungman and J. H. Reibenspies, Dalton Trans., 2009, 8891–8899 RSC.
- T. Sakamoto, K. Watanabe, Y. Shichibu, K. Konishi, S.-I. Sato and T. Nakano, J. Poly. Sci. Poly. Chem., 2011, 49, 945–956 CrossRef CAS.
- M. Suzuki, A. Watanabe, R. Kawai, R. Sato, S.-I. Matsuoka and S. Kawauchi, Polymer, 2021, 215, 123386 CrossRef CAS.
- B. A. Fultz, D. Beery, B. M. Coia, K. Hanson and J. G. Kennemur, Polym. Chem., 2020, 11, 5962–5968 RSC.
- W. Han, C. Luo, Y. Yang, J. Ren, H. Xuan and L. Ge, Colloids Surf., A, 2018, 558, 488–494 CrossRef CAS.
- M. Tu-morn, N. Pairoh, W. Sutapun and T. Trongsatitkul, Mater. Today: Proc, 2019, 17, 2048–2061 CAS.
- J. Salač, J. Šerá, M. Jurča, V. Verney, A. A. Marek and M. Koutný, Materials, 2019, 12, 481 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details; spectroscopic and crystallographic data for 1; SEC traces, spectroscopic and thermal (TGA, DSC) data for the polymers; CCDC 2089243. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2cc01322c |
| This journal is © The Royal Society of Chemistry 2022 |