
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Importance of molecular symmetry for enantiomeric excess recognition by NMR†‡
Karolis
Norvaiša
 a,
John E.
O’Brien
b,
Irina
Osadchuk
c,
Brendan
Twamley
b,
Victor
Borovkov
c and
Mathias O.
Senge
*ad
a,
John E.
O’Brien
b,
Irina
Osadchuk
c,
Brendan
Twamley
b,
Victor
Borovkov
c and
Mathias O.
Senge
*ad
aSchool of Chemistry, Chair of Organic Chemistry, Trinity Biomedical Sciences Institute, 152–160 Pearse Street, Trinity College Dublin, The University of Dublin, Dublin 2, Ireland
bSchool of Chemistry, Trinity College Dublin, The University of Dublin, Dublin 2, Ireland
cDepartment of Chemistry and Biotechnology, Tallinn University of Technology, Akadeemia tee 15, Tallinn 12618, Estonia
dInstitute for Advanced Study (TUM-IAS) Technical University of Munich, Focus Group – Molecular and Interfacial Engineering of Organic Nanosystems, Lichtenbergstrasse 2a, D-85748 Garching, Germany. E-mail: mathias.senge@tum.de; Web: http://www.twitter.com/mathiassenge Web: https://www.sengegroup.eu/
First published on 30th March 2022
Abstract
Recently prochiral solvating agents (pro-CSA) came under the spotlight for the detection of enantiopurity by NMR. Chemical shift non-equivalency in achiral hosts introduced by the presence of chiral guests yields observable resonance signal splitting (Δδ) correlating to the enantiomeric excess (e.e.). In this work, symmetry is our lens to explain porphyrin-based supramolecular receptor activity in a chiral environment. Based on extensive NMR analyses of the atropisomeric receptors, the host symmetry is shown to be affected by porphyrin nonplanarity and further desymmetrized in the presence of a chiral guest. As such, the exposed porphyrin inner core (N–H), with its strong hydrogen bond abilities, for the first time, has been exploited in enantiomeric composition analysis. Our approach in e.e. detection by N–H signals appearing in a previously underutilized region of the spectrum (below 0 ppm) shows chemical shift splitting (Δδ) three times more sensitive to enantiomeric compositions than previously reported systems.
Among the numerous stereodiscrimination methods, nuclear magnetic resonance (NMR) spectroscopy continues to be one of the leading tools for determining the enantiomeric purity of chiral molecules.1 Recently, a new type of NMR spectroscopic detection of enantiomeric excess (e.e.) using prochiral solvating agents (pro-CSA) was introduced by Hill and co-workers.2 In principle, in the event of attractive noncovalent physicochemical interactions, the chiral information of a guest can be transferred to an achiral host and detected as the splitting of the NMR signals. The key example of pro-CSA, N,N′-disubstituted oxoporphyrinogen (Bz2oxP) exhibits a linear response between the e.e. value and the magnitude of β-proton splitting (Δδ) in 1H NMR (Fig. 1a).3 Due to N-alkylation of the Bz2oxP core, the system cannot be protonated and hence suffers serious sensitivity issues compared to unmodified oxP. However, the inevitable prototropic tautomerism and macrocyclic inversions obstruct the potential applications of oxP as a pro-CSA.2a,4 Porphyrins, as prospective pro-CSA candidates for e.e. detection, have also been investigated.5 While 5,10,15,20-tetraphenylporphyrin (TPP) is not affected by the disadvantageous tautomeric processes, as opposed to oxP, the necessary use of low temperatures for the e.e. detection limits the analysis to explicit solvents with a low freezing point (e.g. CDCl3) and analyte solubility during the screening (e.g. precipitation).
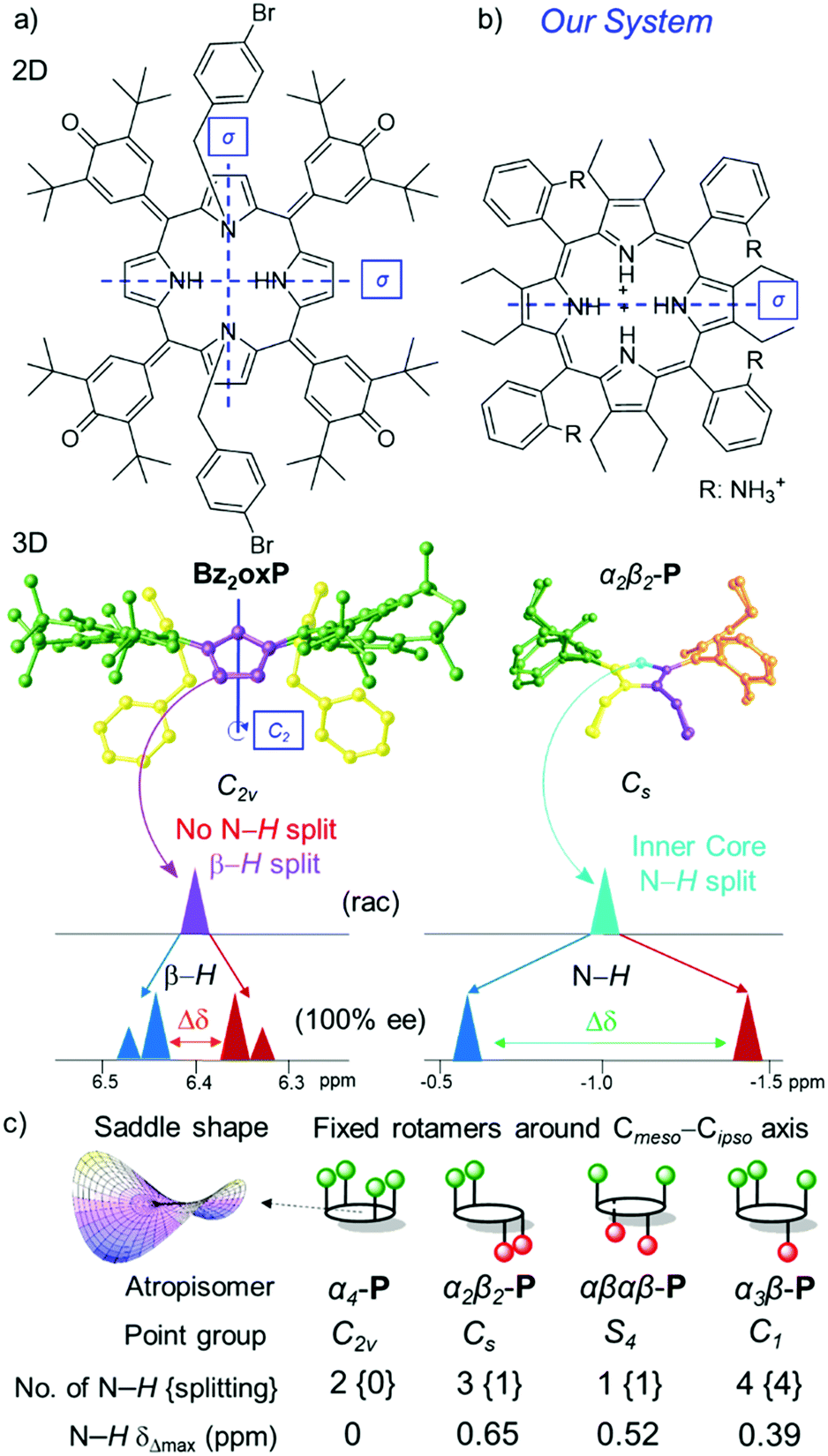 | ||
| Fig. 1 2D (top view) and 3D (side view) representation of pro-CSA's with symmetry elements (mirror plane σ and rotation axis Cn), color coding of symmetrical groups, and the key units used for e.e. detection by 1H NMR in (a) Bz2oxP highlighting β-H splitting;3 (b) newly designed α2β2-P receptor system with chiral discrimination by N–H; (c) All possible P atropisomers with corresponding point groups (note, the symmetry is reduced due to the saddle shape of the macrocycle), N–H signals, and magnitude of splitting; see more detail in Fig. S1 (ESI‡). | ||
Frequently, the use of pro-CSA's 1H NMR spectra for chiral analysis is severely hampered due to the numerous scalar couplings and overlapping signals that lead to analytical difficulties.6 As the majority of organic molecule resonances appear between 0–14 ppm in the 1H NMR scale,7 it is desirable that the e.e. monitoring with pro-CSA would be in a distinct, well-separated region. One of the most unique characteristics of porphyrins is the closed-loop of electrons (ring current) exhibiting large magnetic anisotropy under an applied magnetic field. While peripheral macrocycle signals relate to the typical organic resonances, the nuclei positioned within the loop experience a strong shielding effect when subjected to an external magnetic field and resonate below 0 ppm in the 1H NMR scale.8 Once the highly conjugated system is disrupted (e.g., in oxoporphyrinogens, calix[4]pyrroles), the anisotropic shielding effect of the inner core system is lost, resulting in downfield shifting of the corresponding inner core signals.
The attractive features of the metal-free (free base) porphyrin inner core has lately drawn attention in the fields of catalysis,9 sensing,10 supramolecular assemblies,11 and absolute configuration determination.12 The existing methods of ring puckering by steric strain13 can cause a degree of outwards orientation of the inner pyrrolic entities, making these positions more basic14 and accessible to substrates.10 Even though porphyrins adopt a saddle-shaped 3D conformation15 creating an ‘active center’ in the core, only the saddle-deformation alongside chiral guest interactions is not enough to drive the inner N–H signal to split during the 1H NMR e.e. analysis. For example, Bz2oxP has a saddle shape and belongs to the C2v point-group notation with two mirror planes diagonally dividing all pyrroles (Fig. 1a). The symmetrical nature of Bz2oxP does not permit the e.e. discrimination using the inner core and remain isochronous.3
Here we report the first example of e.e. detection using porphyrin inner core N–H resonances. We have designed P [5,10,15,20-tetrakis(2-aminiumphenyl)-2,3,7,8,12,13,17,18-octaethylporphyrin] as a receptor system (Fig. 2a) exploiting three main molecular engineering strategies: (1) steric overcrowding to obtain a saddle-shaped macrocycle while retaining the porphyrin conjugation13 and exposing the inner pyrrolic units for host–guest interactions; (2) peripheral donating groups creating a lock-and-key10a comparable system to encapsulate chiral analytes in the porphyrin lattice and allow detailed NMR analysis at room temperature;10b (3) formation of atropisomers based on the orientation of peripheral groups16 to have ultimate control of the symmetry elements in pro-CSA.17
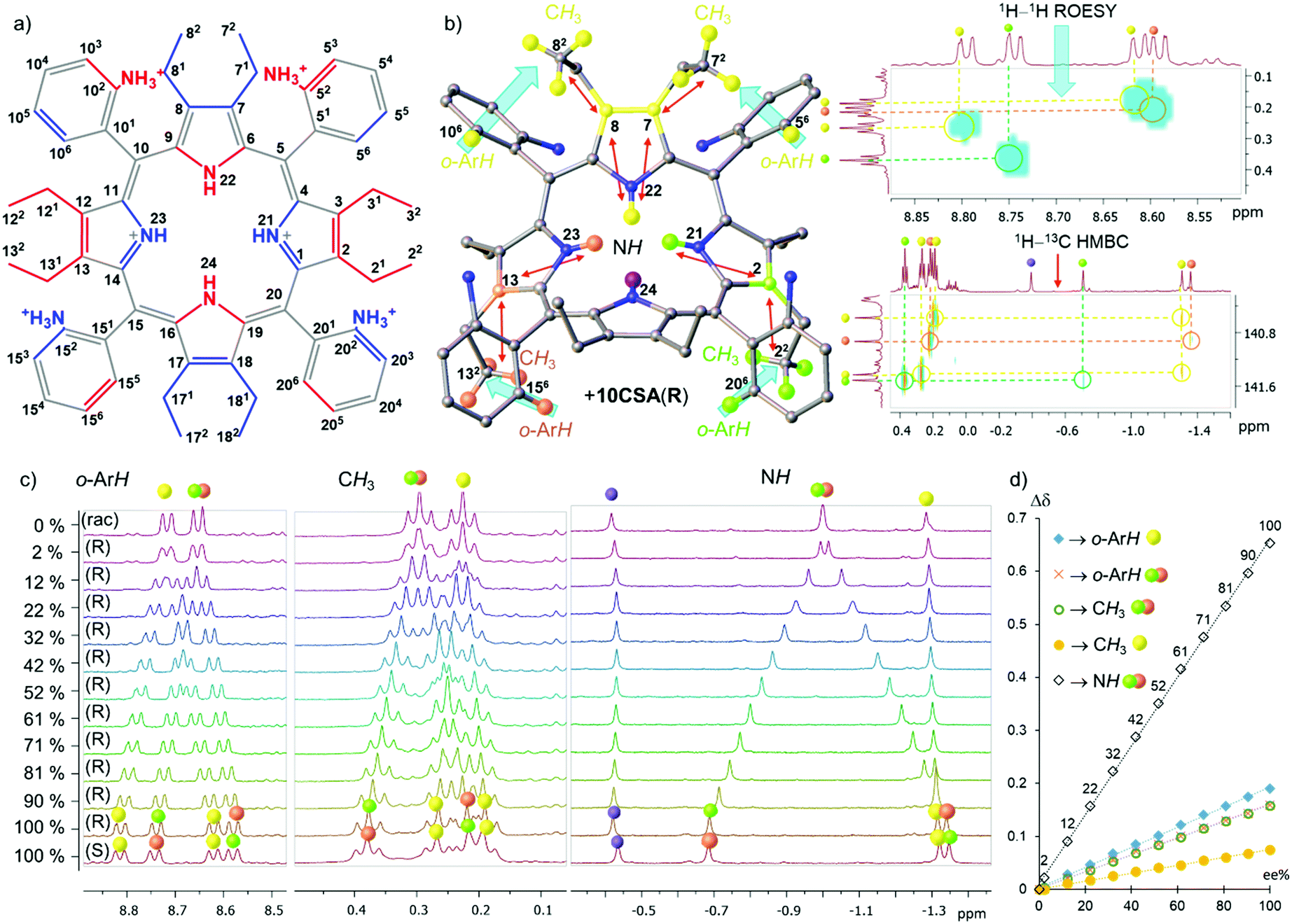 | ||
| Fig. 2 (a) Illustration of the structure of α2β2-P (blue – above and red – below the plane) with corresponding positions; (b) representation and color-coding reference of the 1H splitting signals with blue arrows showing 1H–1H ROESY and red arrows 1H–13C HMBC correlations of 20 eq. α2β2-P·10CSA(R); (c) observable Δσ of 1H signals in o-ArH, CH3, and inner core system (NH) regions; (d) graph of the Δσ dependence on the e.e.% value. All spectra have been recorded in CD3CN. | ||
Previously, we have shown the separation of atropisomers and highlighted selective nature of host P for guests containing sulfonate or phosphonate motifs.10b The analyte interacts directly with the inner ring system and generates static and well-resolved NMR spectral lines.17 As mentioned, low temperatures can also offer slow exchange rates for potential detection of e.e.5 However, the aim of the following studies is the development of a readily available and highly effective analytical tool for room-temperature measurements. Hence, (±)-10-camphorsulfonic acid (10CSA) bearing the sulfonic moiety and stereogenic centers was selected as a chiral guest.
Operating with enantiopure 10CSA(S or R) four distinct scenarios with four different P atropisomers were observed and subsequently rationalized by the symmetry operations found in P (Fig. 1c).17 In the α4-P·10CSA(S or R) complex, the inner core remains isochronous, due to the C2v point-group notation with a two-fold symmetry axis and two mirror planes passing through the pyrroles. The identical situation previously reported by Hill and co-workers in Bz2oxP pinpoints the interactions with inner N–H, however, without the e.e. discrimination due to the C2v symmetry (Fig. 1a).3 The α2β2-P atropisomer with Cs symmetry features a single well-defined mirror plane dividing two pyrrolic units which preserve its achiral nature; hence, allowing it to be classified as pro-CSA. The lack of other symmetry elements in α2β2-P allows the N–H protons to become anisochronous in a chiral environment, making chiral discrimination possible (with the highest magnitude of splitting (Δδmax) of 0.653 ppm at 100% e.e.) (Fig. 1b). The α3β-P atropisomer belongs to the C1 point-group, as it contains no symmetry elements, making the system chiral. Thus, eight signals are observed with enantiopure 10CSA due to diastereomer formation (SS- and SR- or RR- and RS-) (Fig. S1, ESI‡).
While the e.e. detection is possible with α3β-P (Fig. S2, ESI‡), the practical use of such system falls short mainly due to three dominating factors: (1) the high number of inner core system signals hampers direct e.e. interpretation; (2) the magnitude of Δδmax (∼0.39 ppm) is lowest of the three atropisomers with inner core splitting making it the least sensitive system; (3) the concentration of α3β-P is required to be significantly higher than that of other systems due to a large number of resonance signals and their comparatively lower intensities. On the other hand, αβαβ-P which belongs to the S4 point group has four equivalent protons located in the principal axis. While it has no mirror planes, the inversion center situated between the pyrrole units allows the inner core protons to split in equal proportions (above and below the plane) upon interaction with a chiral analyte. A single isochronous N–H signal of αβαβ-P-10CSA(S or R) becomes anisochronous, with Δδmax (0.520 ppm) comparable to the α2β2-P·10CSA(S or R) system (0.653 ppm). While a singular inner core proton splitting is an attractive feature, the practicality of such a system in the e.e. detection is challenging, mainly due to the low atropisomeric rotational barrier, which leads to the formation of other atropisomers at room temperature10b and low abundancy (only 1/8 obtained from statistical mixtures) in comparison to other P rotamers. Since α2β2-P displayed the highest Δσmax value compared to other P atropisomeric species (Fig. 1c), in-depth chirality determination studies listed below were carried out with this receptor system (Fig. 2).
Overall, three distinct and well-resolved regions (o-ArH, CH3, and N–H) were identified for possible e.e. monitoring with α2β2-P (Fig. 2c and Fig. S5, ESI‡). The correlation between the signals of interest was investigated by 2D NMR techniques with enantiopure 10CSA(R) (20 eq.) and their corresponding locations are illustrated in Fig. 2b. The gradual addition of 10CSA(R) to α2β2-P and the influence of water on Δσmax as a competitive agent is detailed in the ESI‡ (Fig. S3–S9). While the Δσmax values of o-ArH and CH3 are comparable to known pro-CSAs2–5,18 being 0.190 ppm (o-ArH yellow), 0.159 ppm (o-ArH red/green), 0.158 ppm (CH3 red/green), and 0.075 ppm (CH3 yellow), the Δσmax values of the inner system (N–H red/green) was found to be more than threefold greater than those of other regions (0.653 ppm).
Since the Δσmax value of the inner core system is substantially higher than that of other regions, the resolution, of which e.e. can be detected, is considerably enhanced. Astonishingly, at as low as 2% e.e., two distinct N–H resonance singlets (Δσ 0.022 ppm.) can clearly be identified, while the other regions show only a broadening of the signals. Plotting the differences in the chemical shifts of split peaks against the % e.e. values revealed a linear dependency with the R2 values being above 0.997 and the inner N–H fitting R2 = 0.9994 (Fig. 2d). The linear fit of the plots is a fundamental property in unlocking the easy calibration of the referenced systems for quick detection of the e.e. value (a detailed example shown in ESI;‡ Fig. S10–S12). Moreover, spatially distant neighboring protons from N–H offer another important feature. Sharp and well-isolated singlets do not suffer from any vicinal scalar J-couplings or roofing effects underlining the simplicity in tracking chiral compositions.
The non-equivalency of α2β2-P·10CSA(S) in 13C and 15N NMRs compared to racemic α2β2-P·10CSA(SR), shows most of the macrocyclic ring system Δσmax > 0.3 ppm with the central two nitrogen atoms having Δσmax = 1.67 ppm (Fig. 3 and Table S1, ESI‡). Nevertheless, due to the greater distance from the active site, most of the phenyl ring resonance signals remained isochronous. Despite this, two particularly different scenarios were portrayed by the o-Ar-13C NMR signals. The Δσmax between 156 and 206 positions yielded excellent separation (∼1.3 ppm), whereas the 56 and 106 imposed only marginal Δσmax (0.04 ppm). A closer examination of the crystal structure of α2β2-P[SO42−][HSO4−]4 revealed a closer distance between C156 and C206 (∼6.429 Å) than between C56 and C106 (∼9.311 Å), subsequently forming a narrow channel for the chiral guest to occupy (Fig. 3). Moreover, the calculated chemical shifts of non-hydrogen atoms in α2β2-P·10CSA(R) using the GIAO-B3LYP/6-311++G**//BP86-D3BJ/def-SVP method and SMD solvent model correlated well with the splitting patterns observed experimentally (see ESI,‡ Table S8). A comparison of the α2β2-P·10CSA(S and SR) splitting resonance signals to other atropisomeric species is detailed in ESI‡ (Tables S2 and S3).
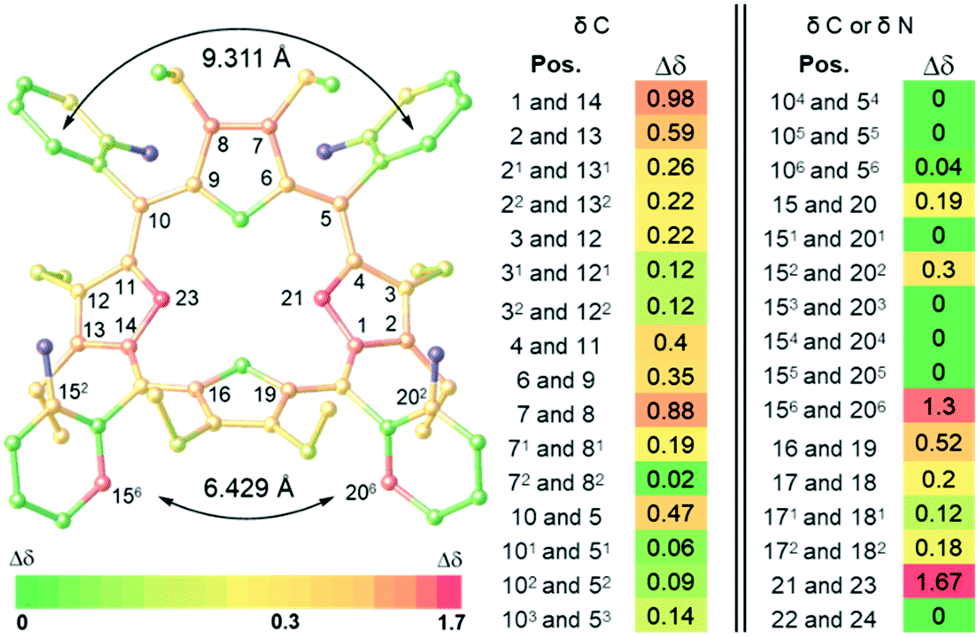 | ||
| Fig. 3 Illustration of Δσmax (ppm) of 13C and 15N NMR in 20 eq. α2β2-P-10CSA(S) complex, determined in comparison to the corresponding racemate α2β2-P-10CSA(SR) using 2D NMR techniques (CD3CN) (Fig. S16–S31, ESI‡). The highlighted positions in the illustration on the left side shows Δσ ≥ 0.3 ppm. Atoms in blue are peripheral nitrogen atoms that did not resonate. | ||
To conclude, the point groups play a fundamental role in adjusting supramolecular receptor systems for e.e. determinations by the NMR method. Four atropisomers containing different point group notations were thoroughly investigated by NMR with (S and R) camphorsulphonic acid pinpointing the α2β2 rotamer as the most sensitive receptor for chirality detection. It was found that the Δσmax value of N–H signals can reach 0.653 ppm, a three-fold greater splitting than any known pro-CSA. Such enhanced sensitivity towards the chiral components allows for readily available and detailed enantiomeric excess detection at room temperature by NMR.
This work was prepared with the support of the Technical University of Munich – Institute for Advanced Study through a Hans Fischer Senior Fellowship and received funding from the European Union's Horizon 2020 research and innovation program under FET-OPEN Grant no. 828779, the Irish Research Council (GOIPG 2017/1172), Science Foundation Ireland (IvP 13/IA/1894), and the Estonian Research Council (Grant PUTJD749 for I. O.). Computations were performed on the HPC cluster of Tallinn University of Technology.
Conflicts of interest
There are no conflicts to declare.References
- (a) Z. Szakács, Z. Sánta, A. Lomoschitz and C. Szántay, Trends Anal. Chem., 2018, 109, 180–197 CrossRef; (b) M. S. Silva, Molecules, 2017, 22, 247–268 CrossRef PubMed; (c) J. S. Fossey, E. V. Anslyn, W. D. G. Brittain, S. D. Bull, B. M. Chapin, C. S. Le Duff, T. D. James, G. Lees, S. Lim, J. A. C. Lloyd, C. V. Manville, D. T. Payne and K. A. Roper, J. Chem. Educ., 2017, 94, 79–84 CrossRef CAS.
- (a) J. Labuta, J. P. Hill, S. Ishihara, L. Hanyková and K. Ariga, Acc. Chem. Res., 2015, 48, 521–529 CrossRef CAS PubMed; (b) A. Shundo, J. Labuta, J. P. Hill, S. Ishihara and K. Ariga, J. Am. Chem. Soc., 2009, 131, 9494–9495 CrossRef CAS PubMed.
- J. Labuta, S. Ishihara, T. Šikorský, Z. Futera, A. Shundo, L. Hanyková, J. V. Burda, K. Ariga and J. P. Hill, Nat. Commun., 2013, 4, 2188–2196 CrossRef PubMed.
- (a) J. Labuta, S. Ishihara, K. Ariga and J. Hill, Symmetry, 2014, 6, 345–367 CrossRef CAS; (b) J. Labuta, Z. Futera, S. Ishihara, H. Kourilova, Y. Tateyama, K. Ariga and J. P. Hill, J. Am. Chem. Soc., 2014, 136, 2112–2118 CrossRef CAS PubMed.
- (a) J. Labuta, S. Ishihara and J. P. Hill, J. Porphyrins Phthalocyanines, 2020, 24, 320–329 CrossRef CAS; (b) J. Labuta, S. Ishihara, A. Shundo, S. Arai, S. Takeoka, K. Ariga and J. P. Hill, Chem. – Eur. J., 2011, 17, 3558–3561 CrossRef CAS PubMed.
- J. Labuta, S. Ishihara, D. T. Payne, K. Takimoto, H. Sato, L. Hanyková, K. Ariga and J. P. Hill, Chemosensors, 2021, 9, 259–276 CrossRef CAS.
- (a) G. R. Fulmer, A. J. M. Miller, N. H. Sherden, H. E. Gottlieb, A. Nudelman, B. M. Stoltz, J. E. Bercaw and K. I. Goldberg, Organometallics, 2010, 29, 2176–2179 CrossRef CAS; (b) M. Balci, in Basic 1H- and 13C-NMR Spectroscopy, ed. M. Balci, Elsevier Science, Amsterdam, 2005, pp. 25–85 Search PubMed.
- J. E. Falk, in Porphyrins and Metalloporphyrins, ed. K. M. Smith, Elsevier Scientific Pub. Co, Amsterdam, 1975, pp. 399–514 Search PubMed.
- M. Roucan, M. Kielmann, S. J. Connon, S. S. R. Bernhard and M. O. Senge, Chem. Commun., 2018, 54, 26–29 RSC.
- (a) M. Kielmann and M. O. Senge, Angew. Chem., Int. Ed., 2019, 58, 418–441 CrossRef CAS PubMed; (b) K. Norvaiša, K. J. Flanagan, D. Gibbons and M. O. Senge, Angew. Chem., Int. Ed., 2019, 58, 16553–16557 CrossRef PubMed; (c) K. Norvaiša, M. Kielmann and M. O. Senge, ChemBioChem, 2020, 21, 1793–1807 CrossRef PubMed.
- (a) C. J. Kingsbury, K. J. Flanagan, H.-G. Eckhardt, M. Kielmann and M. O. Senge, Molecules, 2020, 25, 3195–3218 CrossRef CAS; (b) K. Norvaiša, K. Yeow, B. Twamley, M. Roucan and M. O. Senge, Eur. J. Org. Chem., 2021, 1871–1882 CrossRef.
- P. Bhyrappa, V. V. Borovkov and Y. Inoue, Org. Lett., 2007, 9, 433–435 CrossRef CAS PubMed.
- M. O. Senge, Chem. Commun., 2006, 243–256 RSC.
- O. S. Finikova, A. V. Cheprakov, P. J. Carroll, S. Dalosto and S. A. Vinogradov, Inorg. Chem., 2002, 41, 6944–6946 CrossRef CAS PubMed.
- C. J. Kingsbury and M. O. Senge, Coord. Chem. Rev., 2021, 431, 213760 CrossRef CAS.
- K. Norvaiša, S. Maguire, C. Donohoe, J. E. O'Brien, B. Twamley, L. C. Gomes-da-Silva and M. O. Senge, Chem. – Eur. J., 2022, 28, e202103879 CrossRef PubMed.
- K. Norvaiša, J. E. O'Brien, D. J. Gibbons and M. O. Senge, Chem. – Eur. J., 2020, 27, 331–339 CrossRef.
- (a) S. Ishihara, J. Labuta, Z. Futera, S. Mori, H. Sato, K. Ariga and J. P. Hill, J. Phys. Chem. B, 2018, 122, 5114–5120 CrossRef CAS PubMed; (b) K. Takimoto, S. Ishihara, J. Labuta, V. Březina, D. T. Payne, J. P. Hill, K. Ariga, M. Sumita, S. Mori and H. Sato, J. Phys. Chem. Lett., 2020, 11, 8164–8169 CrossRef CAS PubMed.
Footnotes |
| † A previous version of this manuscript has been deposited on a preprint server (https://doi.org/10.26434/chemrxiv-2022-xg9bd). |
| ‡ Electronic supplementary information (ESI) available: CCDC 2143572 (for α2β2-P[SO42−][HSO4−]4). For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2cc01319c |
| This journal is © The Royal Society of Chemistry 2022 |