
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Extension of hydrogen borrowing alkylation reactions for the total synthesis of (−)-γ-lycorane†
Christopher J. J.
Hall
a,
Indi S.
Marriott
a,
Kirsten E.
Christensen‡
a,
Aaron J.
Day
a,
William R. F.
Goundry
b and
Timothy J.
Donohoe
 *a
*a
aDepartment of Chemistry, University of Oxford, Chemistry Research Laboratory, Mansfield Road, Oxford, OX1 3TA, UK. E-mail: timothy.donohoe@chem.ox.ac.uk
bEarly Chemical Development, Pharmaceutical Sciences, R&D, AstraZeneca, Macclesfield, UK
First published on 29th March 2022
Abstract
The total synthesis of (–)-γ-lycorane (10 steps) and synthesis of (±)-γ-lycorane (8 steps) was completed from cyclohexenone. A new two step hydrogen borrowing alkylation of an aziridinyl alcohol, coupled with a Ph* (Me5C6) deprotection/cyclisation procedure was developed for de novo formation of the fused 6,5 heterocyclic ring. This work is one of the first examples of hydrogen borrowing C–C bond formation being used as a key step in a total synthesis project.
The development of hydrogen borrowing catalysis has given rise to several novel C–C bond forming methodologies.1 The primary strategic advantage of a hydrogen bonding approach for the C-alkylation of enolates is that it allows the direct use of alcohols as alkylating reagents, without the need for a formal activation step (e.g. conversion to halide or pseudo-halide).2 Recently, we developed the use of chiral 1,2-aminoalcohols as novel alkylating agents that can be added as electrophiles in hydrogen borrowing C–C bond forming catalysis.3 As our next objective, we sought to expand on the utilisation of the 1,2-aminoalcohol motif in the context of a chemical synthesis project. Our attention was drawn to the possible use of 1,2-aziridinyl alcohols (especially cyclic compounds such as A, Scheme 1) as alkylating agents. As a general strategy, the successful alkylation of a methyl ketone (here Ph*COMe)4 with the general alcohol structure as A would facilitate some intriguing possibilities for further elaboration. If we could open the product aziridine B regio- and stereoselectively to form C then deprotection of the Ph* group would allow the possibility of cyclisation to form a cis-fused 6,5-lactam system D that is found in many natural products either as the lactam or fully reduced form (see 1 and 2, Scheme 1).
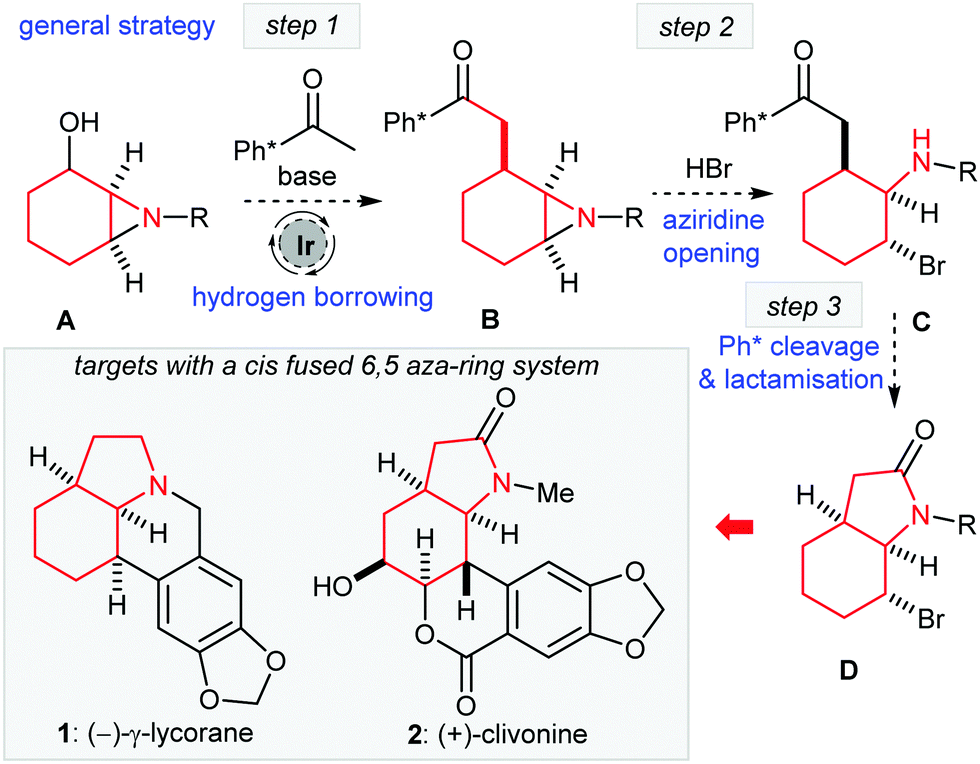 | ||
| Scheme 1 Hydrogen borrowing alkylation of aziridinyl alcohols and a route to 6,5-fused ring systems: Ph* = Me5C6. | ||
We decided to test our hypotheses regarding aziridinyl alcohol alkylation and ring opening in the context of a synthesis of γ-lycorane (1). This is an interesting target for total synthesis for several reasons. Firstly, the pentacyclic structure with three contiguous cis stereocentres provides a challenging target for hydrogen borrowing methodology. Moreover, the asymmetric aziridination of cyclohexenone could be recruited to allow the preparation of enantiopure A for subsequent hydrogen borrowing alkylation; this should then allow the preparation of enantiopure lycorane.5 Note that γ-lycorane (1) itself is not thought to be a natural product, but a degradation product of several members of the caranine family of alkaloids, first reported by Kotera in 1961,6 and it has proven to be a popular target for total synthesis.7
To test our strategy for the asymmetric synthesis of (–)-1, aziridine 5 was prepared from cyclohexanone (3) in 98![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 er, by following a modified procedure of Hamada and co-workers using chiral diamine 4 (Scheme 2).8 Reduction of the ketone 5 gave aziridyl alcohol 6 with modest diastereoselectivity. Next, the Cbz aziridine was smoothly converted into the piperonyl amine (8) in two steps comprising of Cbz removal and N-alkylation. Then we were ready to test the first key step, namely hydrogen borrowing catalysed alkylation of Ph*COMe with aziridinyl alcohol 8 (note that this class of alcohol has not been previously employed in hydrogen borrowing alkylation). Pleasingly, the iridium catalysed alkylation of 9 with (+)-α-aziridyl alchohol 8 afforded 11 in 53% yield as a single (all cis) diastereoisomer.9 We presume that the desired cis stereochemistry of 11 derives from selective [Ir–H] reduction of enone intermediate 10 from the less hindered convex face.
:2 er, by following a modified procedure of Hamada and co-workers using chiral diamine 4 (Scheme 2).8 Reduction of the ketone 5 gave aziridyl alcohol 6 with modest diastereoselectivity. Next, the Cbz aziridine was smoothly converted into the piperonyl amine (8) in two steps comprising of Cbz removal and N-alkylation. Then we were ready to test the first key step, namely hydrogen borrowing catalysed alkylation of Ph*COMe with aziridinyl alcohol 8 (note that this class of alcohol has not been previously employed in hydrogen borrowing alkylation). Pleasingly, the iridium catalysed alkylation of 9 with (+)-α-aziridyl alchohol 8 afforded 11 in 53% yield as a single (all cis) diastereoisomer.9 We presume that the desired cis stereochemistry of 11 derives from selective [Ir–H] reduction of enone intermediate 10 from the less hindered convex face.
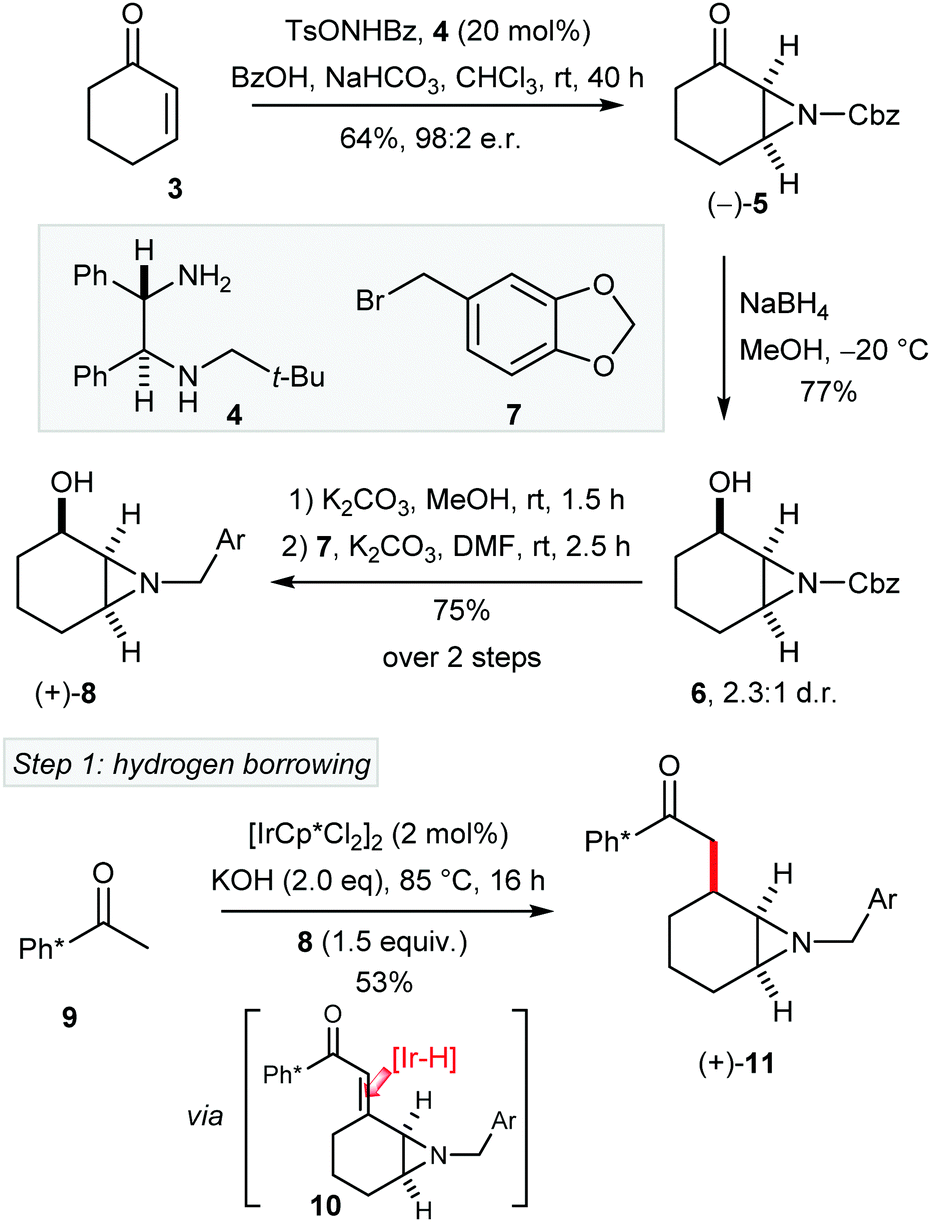 | ||
| Scheme 2 Asymmetric synthesis of fragment 11. Er of 5 measured by HPLC against a racemic standard. | ||
At the same time as our development of a route to enantioenriched (–)-γ-lycorane, a route to (±)-1 was also completed (Scheme 3). In this case the lack of requirement for an asymmetric aziridination allowed access to 11via a shorter sequence. Thus, aziridination of 12 (prepared from cyclohexanone in one step)10 with piperonylamine (13), and reduction of the resultant ketone 14 gave the α-aziridyl alchohol (±)-8 as a single diastereoisomer in excellent yield. Next, racemic 8 was used to alkylate Ph* methyl ketone (9) affording (±)-11 in 60% yield. However, in this route we questioned the need to reduce ketone 14 to alcohol 8, only to have it re-oxidised in the hydrogen borrowing step. To this end, we envisioned the direct alkylation of ketone 9 with another ketone (here 14). Note that this particular reaction necessitates the addition of a stoichiometric hydrogen donor to provide hydride for reduction of the enone precursor to 11 (i.e. (±)-10). After experimentation, we selected alcohol 1511 which we reasoned would be readily oxidised in situ, and thus provide hydride for the catalyst controlled enone reduction. Note that in this case the ketone by-product from this oxidation would not readily compete in the aldol reactions that occur in hydrogen borrowing alkylation. Pleasingly, the use of the benzhydrol derivative 15 gave (±)-11 directly from ketone (±)-14 with only a slightly diminished yield compared to the two-step procedure.
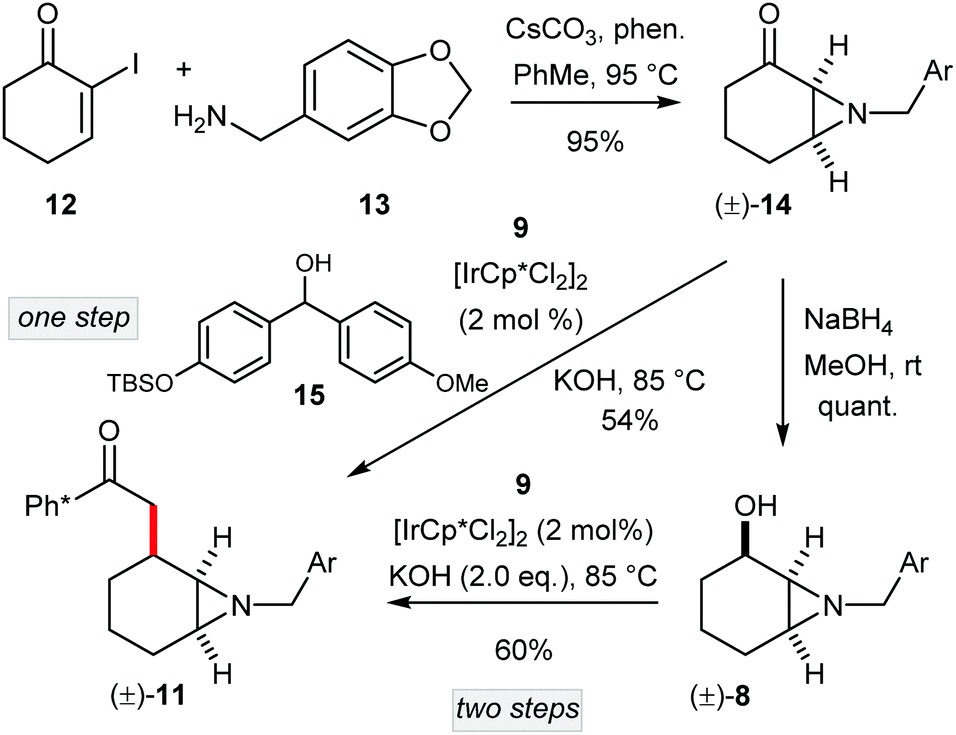 | ||
| Scheme 3 Synthesis of racemic fragment 11. | ||
With both enantiopure and racemic 11 in hand we now turned to elaboration of the aziridine and completion of the synthesis (Scheme 4). Pleasingly, preliminary experiments had shown that cleavage of the Ph* group from 11 using molecular bromine had the beneficial added effect of activating the acyl carbonyl as either an acylium ion or acid bromide. We found that the aziridine nitrogen was able to intercept this reactive intermediate to form an aziridinium ion in situ; this was subsequently opened regio- and stereoselectively by a strain-release SN2 displacement by bromide ion. Thus, this protocol allowed the combination of steps 2 and 3 from the general plan (Scheme 1).
 | ||
| Scheme 4 Endgame for the synthesis of γ-lycorane 1. Yields in parentheses are for racemic material. Er of 1 was measured by HPLC against a racemic standard. | ||
Treatment of 11 with Br2 provided smooth conversion to the dibrominated lactam 16 in 44% yield (78% for racemic 11). Note that this reaction also delivered, as desired, a mono-bromination of the aromatic ring. From here, regioselective elimination of the alkyl bromide followed by a regio- and stereoselective intramolecular Heck reaction forged the final C–C bond to give 17 in 67% yield (60% racemic). Finally, catalytic hydrogenation of the alkene with Pd/C, followed by amide reduction with LiAlH4 afforded (–)-γ-lycorane (1) in 68% yield (66% racemic). The spectroscopic data for the synthetic material matched that reported in the literature, and we were also able to obtain a single crystal X-ray diffraction structure of racemic lycorane which confirmed the relative stereochemistry of this product.12
In summary we have completed the synthesis of (−)-lycorane in 10 steps and of (±)-lycorane in 8 steps. The hydrogen borrowing alkylation of an azridinyl alcohol was crucial in our synthetic strategy, and this general methodology should allow the synthesis of a broad range of complex polycyclic nitrogen containing natural products. Furthermore, to the best of our knowledge, this is the first case where C–C bond forming hydrogen borrowing catalysis has been employed as a key step in a total synthesis. Future work will concentrate on expanding the applicability of hydrogen borrowing strategies in chemical synthesis projects.
We thank the EPSRC (grant EP/T011599/1, AJD). C. J. J. H. is grateful to the EPSRC Centre for Doctoral Training in Synthesis for Biology and Medicine (EP/L015838/1) for a studentship, generously supported by AstraZeneca, Diamond Light Source, Defence Science and Technology Laboratory, Evotec, GlaxoSmithKline, Janssen, Novartis, Pfizer, Syngenta, Takeda, UCB and Vertex.
Conflicts of interest
There are no conflicts to declare.Notes and references
- For recent reviews on hydrogen borrowing catalysis see: (a) J. M. Ketcham, I. Shin, T. P. Montgomery and M. J. Krische, Angew. Chem., Int. Ed., 2014, 126, 9294 CrossRef; (b) H. Aitchison, R. L. Wingad and D. F. Wass, ACS Catal., 2016, 6, 7125 CrossRef CAS; (c) M. Holmes, L. A. Schwartz and M. J. Krische, Chem. Rev., 2018, 118, 6026 CrossRef CAS PubMed; (d) B. G. Reed-Berendt, K. Polidano and L. C. Morrill, Org. Biomol. Chem., 2019, 17, 1595 RSC; (e) T. Kwok, O. Hoff, R. J. Armstrong and T. J. Donohoe, Chem. – Eur. J., 2020, 26, 12912 CrossRef CAS PubMed; (f) C. Gazolla Santana and M. J. Krische, ACS Catal., 2021, 11, 5572 CrossRef PubMed.
- (a) C. J. J. Hall, W. R. F. Goundry and T. J. Donohoe, Angew. Chem., Int. Ed., 2021, 60, 69811 CrossRef PubMed . See also: ; (b) S. Bartolucci, M. Mari, A. Bedini, G. Piersanti and G. Spandoni, J. Org. Chem., 2015, 80, 3217 CrossRef CAS PubMed; (c) S. Bartolucci, M. Mari, G. Di Gregorio and G. Piersanti, Tetrahedron, 2016, 72, 2233 CrossRef CAS; (d) G. Di Gregorio, M. Mari, S. Bartolucci, F. Bartolucci and G. Piersanti, Org. Chem. Front., 1622, 2018, 5 Search PubMed; (e) F. Bartoccini, M. Retini and G. Piersanti, Tetrahedron Lett., 2020, 61, 151875 CrossRef CAS; (f) A. E. R. Chamberlain, K. J. Paterson, R. J. Armstrong, H. C. Twin and T. J. Donohoe, Chem. Commun., 2020, 56, 3563 RSC.
- See for example: (a) S. Elangovan, J.-B. Sortais, M. Beller and C. Darcel, Angew. Chem., Int. Ed., 2015, 54, 14483 CrossRef CAS PubMed; (b) N. Diebl and R. Kempe, J. Am. Chem. Soc., 2016, 138, 10786 CrossRef PubMed; (c) W. M. Akhtar, C. B. Cheong, J. R. Frost, K. E. Christensen, N. G. Stevenson and T. J. Donohoe, J. Am. Chem. Soc., 2017, 139, 2577 CrossRef CAS PubMed; (d) W. M. Akhtar, R. J. Armstrong, J. R. Frost, N. G. Stevenson and T. J. Donohoe, J. Am. Chem. Soc., 2018, 140, 11916 CrossRef CAS PubMed; (e) L. B. Smith, R. J. Armstrong, D. Matheau-Raven and T. J. Donohoe, J. Am. Chem. Soc., 2020, 142, 2514 CrossRef CAS PubMed; (f) M. B. Dumbatta, J. Santos, R. R. A. Bolt and L. C. Morill, Tetrahedron, 2020, 76, 131571 CrossRef.
- For a review see: R. J. Armstrong and T. J. Donohoe, Tetrahedron Lett., 2021, 74, 153151 CrossRef CAS.
- For a review of asymmetric aziridination reactions see: L. Degennaro, P. Trinchera and R. Luisi, Chem. Rev., 2014, 114, 7881 CrossRef CAS PubMed.
- K. Kotera, Tetrahedron, 1961, 12, 248 CrossRef CAS.
- For previous syntheses of γ-lycorane (1) see: (a) H. Iida, Y. Yuasa and C. Kibayashi, J. Am. Chem. Soc., 1978, 100, 3598 CrossRef CAS; (b) J.-E. Backvall, P. G. Andersson, G. B. Stone and A. Gogoll, J. Org. Chem., 1991, 56, 2988 CrossRef; (c) W. H. Pearson and J. M. Sckeryantz, J. Org. Chem., 1992, 57, 6783 CrossRef CAS; (d) H. Yoshizaki, H. Satoh, Y. Sato, S. Nukui, M. Shibasaki and M. Mori, J. Org. Chem., 1995, 60, 2016 CrossRef CAS; (e) J. Cossy, L. Tresnard and D. G. Pardo, Eur. J. Org. Chem., 1999, 1925 CrossRef CAS; (f) M. G. Banwell, J. E. Harvey, D. C. R. Hockless and A. W. Wu, J. Org. Chem., 2000, 65, 4241 CrossRef CAS PubMed; (g) A. Padwa, M. A. Brodney and S. M. Lynch, J. Org. Chem., 2001, 66, 1716 CrossRef CAS PubMed; (h) L. Dong, T.-J. Xu, L.-F. Cun, X. Cui, A.-Q. Mi, Y.-Z. Jiang and L.-Z. Gong, Org. Lett., 2005, 7, 4285 CrossRef CAS PubMed; (i) S. Gao, Y. Q. Tu, Z. Song, A. Wang, X. Fan and Y. Jiang, J. Org. Chem., 2005, 70, 6524 Search PubMed; (j) B. D. Chapsal and I. Ojima, Org. Lett., 2006, 8, 1395 CrossRef CAS; (k) H. Fujioka, K. Murai, Y. Ohba, H. Hirose and Y. Kita, Chem. Commun., 2006, 832 RSC; (l) S. A. A. El Bialy, Nat. Prod. Res., 2008, 22, 1176 CrossRef CAS PubMed; (m) E. S. Conner, K. E. Crocker, R. G. Fernando, F. R. Fronczek, G. G. Stanley and J. R. Ragains, Org. Lett., 2013, 15, 5558 CrossRef CAS PubMed; (n) D. Liu, L. Ai, F. Li, A. Zhao, J. Chen, H. Zhang and J. Liu, Org. Biomol. Chem., 2014, 12, 3191 RSC; (o) I. A. Andreev, N. K. Ratmanova, A. M. Novoselov, D. S. Belov, I. F. Seregina and A. V. Kurkin, Chem. – Eur. J., 2016, 22, 7262 CrossRef CAS PubMed; (p) A. Monaco, B. R. Szulc, Z. X. Rao, M. Barniol-Xicota, M. Sehailia, B. M. A. Borges and S. T. Hilton, Chem. – Eur. J., 2017, 23, 4750 CrossRef CAS PubMed; (q) W. L. Yu, T. Nunns, J. Richardson and K. I. Booker-Milburn, Org. Lett., 2018, 20, 1272 CrossRef CAS PubMed; (r) B. N. D. Doan, X. Y. Tan, C. M. Ang and R. W. Bates, Synthesis, 2017, 4711 CAS; (s) R. Rocaboy, D. Dailler and O. Baudoin, Org. Lett., 2018, 20, 772 CrossRef CAS PubMed; (t) Y. Pang, H. Xiao, W. Ou, X. Zhang, X. Wang and S. Huang, Tetrahedron Lett., 2020, 61, 151733 CrossRef CAS; (u) K. B. Wilson, H. S. Nedzbala, S. R. Simpson, M. N. Ericson, K. S. Westendorff, M. D. Chordia, D. A. Dickie and W. D. Harman, Helv. Chim. Acta, 2021, 104, e2100103 CrossRef CAS PubMed.
- Y. Menjo, A. Hamajima, N. Sasaki and Y. Hamada, Org. Lett., 2011, 13, 5744 CrossRef CAS PubMed.
- The major all cis isomer of 8 was used in the alkylation step as the reaction give higher yield compared to use of the 2.3:1 diastereomeric mixture.
- M. E. Krafft and J. W. Cran, Synlett, 2005, 1263 CrossRef CAS.
- T. Misawa, H. Aoyama, T. Furuyama, K. Dodo, M. Sagawa, H. Miyachi, M. Kizaki and Y. Hashimoto, Chem. Pharm. Bull., 2008, 56, 1490 CrossRef CAS.
- For details concerning solving and refining this structure, see: (a) L. Palatinus and G. Chapuis, J. Appl. Crystallogr., 2007, 40, 786–790 CrossRef CAS; (b) P. Parois, R. I. Cooper and A. L. Thompson, Chem. Cent. J., 2015, 9, 30 CrossRef; (c) R. I. Cooper, A. L. Thompson and D. J. Watkin, J. Appl. Crystallogr., 2010, 43, 1100–1107 CrossRef CAS . CCDC 2151105†.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental procedures, spectroscopic data and copies of NMR spectra are available. CCDC 2151105. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d2cc01248k |
| ‡ Author to whom correspondence regarding the X-ray crystal structure should be addressed. |
| This journal is © The Royal Society of Chemistry 2022 |