
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Stereospecific reaction of sulfonimidoyl fluorides with Grignard reagents for the synthesis of enantioenriched sulfoximines†
Stephanie
Greed
 ,
Oliver
Symes
and
James A.
Bull
*
,
Oliver
Symes
and
James A.
Bull
*
Department of Chemistry, Imperial College London, Molecular Sciences Research Hub, White City Campus, Wood Lane, London W12 0BZ, UK. E-mail: j.bull@imperial.ac.uk
First published on 7th April 2022
Abstract
Sulfonimidoyl halides have previously shown poor stability and selectivity in reaction with organometallic reagents. Here we report the preparation of enantioenriched sulfonimidoyl fluorides and their stereospecific reaction at sulfur with Grignard reagents. Notably the first enantioenriched alkyl sulfonimidoyl fluorides are prepared, including methyl. The nature of the N-group is important to the success of the stereocontrolled sequence to sulfoximines.
Aza-sulfur (VI) derivatives are increasingly validated in drug discovery,1,2 and have seen a marked increase in their use. Sulfoximine containing compounds in particular have entered clinical trials including roniciclib (Bayer)3 and ceralasertib (AstraZeneca).4,5 It is notable that these sulfoximine derivatives, which are chiral at sulfur, are single stereoisomers. In comparison to sulfones, the additional N-vector in sulfoximines provides potential as a H-bond donor, for functionalization or to tune properties.6 Methods for their enantiocontrolled synthesis are of particular value, to exploit the directional nature of potential interactions.
Methods to prepare sulfoximines have seen significant developments in recent years,6,7 including facile methods for NH transfer,8,9 and new reagents containing the SON motif.10,11 Pre-formed methyl sulfoximine reagents have recently been demonstrated to undergo SNAr reactions with heteroarenes.12 However, there remain very few methods for the stereocontrolled construction of sulfoximine derivatives through S–C bond formation. Maruoka has recently reported powerful nucleophilic reagents for sulfoximine synthesis utilising t-butylsulfinamide as a chiral framework.13
Electrophilic reagents to form sulfoximines have been historically challenging, and there are few examples that can provide an enantioenriched product. Early examples of non-racemic sulfonimidoyl chlorides were reported by Cram14a and Johnson,14b but reaction with organometallic reagents resulted in attack at chlorine and reduction to the sulfinamide (e.g. with Grignard reagents, Fig. 1b). Johnson later found that racemic sulfonimidoyl fluorides could be reacted with a limited range of organolithium reagents to form sulfoximines (Fig. 1c).15,16 More recently, Sharpless demonstrated the reaction of phenyl-sulfonimidoyl fluorides with organolithium reagents.11 Notably, all examples to date were N-alkyl or aryl derivatives that were not readily removable to unveil the NH-sulfoximine.
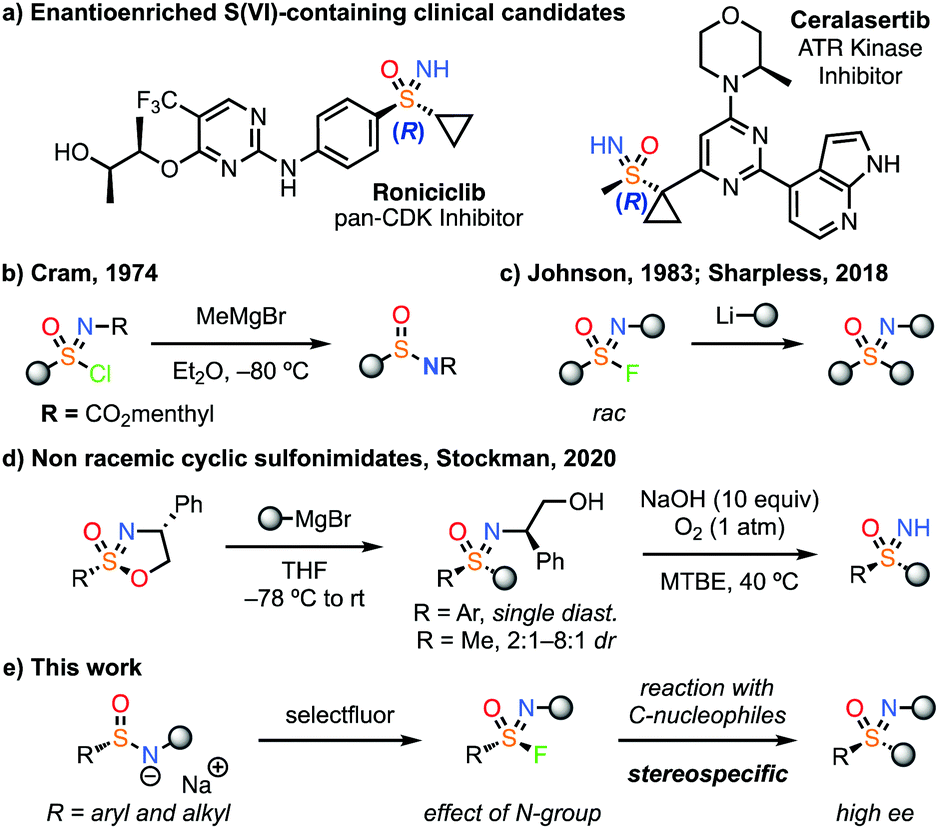 | ||
| Fig. 1 Clinical candidates containing stereochemically pure sulfoximines and electrophilic reagents for sulfoximine synthesis. | ||
To date, the most effective electrophilic reagents to form non-racemic sulfoximines have been cyclic sulfonimidates bearing a chiral auxiliary on nitrogen.17 Building on the work of Reggelin,17a Stockman recently developed cyclic sulfonimidates as separable diastereomers at sulfur, which reacted with Grignard reagents (Fig. 1d).17b Stereospecific conversion was achieved for phenyl sulfonimidates whereas methyl derivatives gave low stereocontrol, leading to mixtures of S-stereoisomers, likely via an initial elimination. The auxiliary was removed using O2 and base.17b
Here we report the generation of highly enantioenriched aryl and alkyl sulfonimidoyl fluorides and their stereospecific reaction to generate sulfoximines by S–C bond formation (Fig. 1e). A broad range of Grignard reagents and other organometallic species were successful to generate highly enantioenriched sulfoximines. Notably, an enantiopure methyl sulfonimidoyl fluoride reagent reacted without loss of ee.
The first reports of enantioenriched sulfonimidoyl fluorides were in 2020 from ourselves18 and Zuilhof19 for reaction with amines and phenolates respectively. Enantioenriched sulfonimidoyl fluorides present interesting potential as synthetic intermediates, and in chemical biology20 and polymer science.21 Fluoride ions were found to cause racemisation of the sulfonimidoyl fluorides through a degenerate exchange,22 which could be avoided by their sequestration.18,21 Aiming to prepare sulfoximines, we investigated the reaction of sulfonimidoyl fluorides with carbon nucleophiles. Despite little encouragement from the literature, we prioritised Grignard reagents as they are widely available and less basic than organolithium reagents. We anticipated that the magnesium halide counterion could scavenge fluoride and prevent fluoride-mediated racemisation of the sulfonimidoyl fluoride.
Initially we investigated NBoc-tolylsulfonimidoyl fluoride 1, prepared at high ee by our previously reported electrophilic fluorination of sulfinamide salts.18 On reaction with 4-methyoxyphenylmagnesium bromide (PMPMgBr) we were delighted to observe that substitution occurred successfully, to give sulfoximine 2a in 58% yield with only a small loss of ee (Table 1, entry 1). The addition of lithium salts (LiCl or LiBr) as had been useful previously with amine nucleophiles18 saw a small increase in es but was detrimental to conversion (Entry 2). Changing the solvent to Et2O was beneficial to both yield and ee (entries 3 and 4) and varying the concentration did not have a significant effect (entries 4–7). Decreasing the equivalents of Grignard reagent and reducing the reaction time to 1 h resulted in a 91% yield (by 1H NMR) and complete retention of ee (entry 9). By comparison, the use of the organolithium reagent (PMPLi) in THF was also successful in retaining the ee, but with significantly reduced yield (entry 10). Using the potentially more functional group tolerant organocuprate reagent, formed from the Grignard reagents as PMP2Cu(MgBr), gave both high ee and 81% isolated yield of 2a in an extended reaction time at rt (entry 11).23 Organozinc reagents were not sufficiently reactive, and returned the sulfonimidoyl fluoride.23
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | [M] | R-[M] equiv. | Solvent (conc.) | Yielda | es (%)b | |
| Reactions performed on 0.10 mmol scale.a Calculated by 1H NMR spectroscopy using 1,3,5-trimethoxybenzene as internal standard.b es determined by HPLC analysis of crude reaction product.c Lithium bromide (1.5 equiv.) added.d Reaction time 1 h.e Addition of organolithium at −78 °C followed by warming to 0 °C.f Reaction time 5 h at rt; 0.25 mmol scale.g Isolated yield.h Reaction time 3 h at rt. | ||||||
| 1 | 2a | |||||
| 1 | MgBr | 1.5 | THF (0.3 M) | 31 | 58 | 98 |
| 2c | MgBr | 1.5 | THF (0.3 M) | 62 | 23 | >99 |
| 3 | MgBr | 1.5 | 1,4-Dioxane (0.3 M) | 50 | 39 | 98 |
| 4 | MgBr | 1.5 | Et2O (0.3 M) | 5 | 77 | 99 |
| 5 | MgBr | 1.5 | Et2O (0.1 M) | — | 70 | 97 |
| 6 | MgBr | 1.5 | Et2O (0.2 M) | — | 69 | 99 |
| 7 | MgBr | 1.5 | Et2O (0.5 M) | — | 70 | 98 |
| 8 | MgBr | 1.2 | Et2O (0.3 M) | — | 81 | 99 |
| 9d | MgBr | 1.2 | Et2O (0.3 M) | — | 91 | >99 |
| 10e | Li | 1.2 | THF (0.3 M) | — | 37 | >99 |
| 11f | [CuAr] | 1.2 | Et2O (0.3 M) | — | 87 (81)g | >99 |
| 12h | ZnCl | 1.2 | Et2O (0.3 M) | 90 | 0 | n/a |

Using these optimised conditions (Table 1, entry 9) gave excellent isolated yields for both the fluorination (1)18 and Grignard reaction (2a obtained in 96% isolated yield and 99% ee on 0.25 mmol scale; Scheme 1). Furthermore, performing the reaction in cyclopentyl methyl ether (CPME) in place of Et2O gave the same result, providing quantitative yield and complete enantiospecificity for 2a in an industrially preferred solvent.
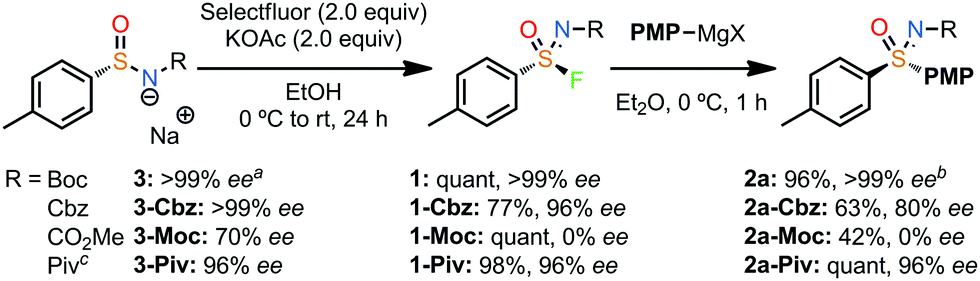 | ||
| Scheme 1 Effect of nitrogen protecting group on the enantiospecificity of the fluorination/Grignard reaction sequence. Fluorination reactions carried out on 0.50 mmol scale. Grignard reaction carried out on 0.25 mmol scale. All yields and %ee values are of isolated product. a ee of the salts recorded after reprotonation. b Comparable result obtained using CPME as solvent (See SI for further details). c Sequence also performed with the opposite enantiomer with comparable results. | ||
The choice of N-group proved to be critical to the success of the sequence. While the NBoc derivative gave excellent enantiospecificity, the same sequence with NCbz gave reduced yields for each step, and a noticeable loss of ee in the Grignard reaction (2a-Cbz). The methyl carbamate was highly susceptible to racemisation, presumably due to reduced steric protection. The NPiv group performed similarly to the Boc group across the sequence retaining very high ee (2a-Piv).
The reaction of the NBoc-tolylsulfonimidoyl fluoride 1 was then explored with a wide variety of Grignard reagents to rapidly prepare a collection of highly enantioenriched sulfoximines (Scheme 2).24 Grignard reagents were used as supplied or prepared by halogen exchange with iPrMgCl·LiCl, which gave comparable results.23 Aryl Grignard reagents gave sulfoximines 2b–2h in excellent yields and enantiospecificity for electron-rich and electron-poor reagents. Notably, these enantioenriched sulfoximines could not be accessed using oxidation/imidation approaches where chiral catalysts would be required to distinguish between electronically and sterically similar substituents on either side of the sulfur atom. Heteroaromatic Grignard reagents derived from thiophene, NBoc-indole and pyridine gave an excellent yield and ee (2i–2k). 2-Methyl-1-propenylmagnesium bromide gave enantiopure vinyl sulfoximine 2l. Allyl and benzyl Grignard reagents were also successful (2m and 2n). Finally, alkyl Grignard reagents, including methyl and cyclopropyl derivatives, gave aryl–alkyl sulfoximines in high yields and with excellent ee (2o–2r). The tolyl methyl sulfoximine derivative 2p allowed confirmation of the stereochemical outcome, by comparison with a known compound.9b,23 This indicated the substitution reaction proceeded with inversion, consistent with an SN2 process. Lithium α-anions of sulfones and sulfoximines were also successfully reacted with 2b with retention of ee.23
 | ||
| Scheme 2 Reaction scope varying the Grignard reagents with sulfonimidoyl fluoride 1. a ee not recorded as separation of enantiomers by HPLC not achieved. | ||
Next, the sulfonimidoyl fluoride was varied (Scheme 3). The 4-bromophenyl sulfonimidoyl fluoride, which was prepared in high ee,18 gave sulfoximine 4 enantiospecifically on reaction with PMPMgBr. Notably, Br–Mg exchange was not observed, retaining a handle for further functionalisation, and providing another advantage of the Grignard reagents over organolithium reagents. Varying the aryl group in a racemic series of sulfonimidoyl fluorides, including pyridine derivatives gave good yields (5–9). Pleasingly, the NBoc-methylsulfonimidoyl fluoride gave a high yield using 1.2 equiv. of the Grignard reagent (10). The reaction also worked well with the iPr derivative (11), however, tBu derivative 12 did not form. The unreactive nature of the tBu-sulfonimidoyl fluoride is consistent with the required nucleophile approach trajectory for SN2. Additional methyl and benzyl derivatives were also demonstrated (13–15).
 | ||
| Scheme 3 Variation of sulfonimidoyl fluoride. | ||
The preparation of enantioenriched sulfinamide derivatives remains challenging and there is very limited commercial availability. Previously we reported an enantioselective oxidation–imination–elimination sequence for 4-bromophenyl-sulfonimidoyl fluoride.18 However, this is much less viable for alkyl derivatives. As such, we turned to the powerful recent reports from Maruoka for the preparation of sulfinamides and sulfoximines, starting from t-butylsulfinamide which is readily available in both enantiomers (Scheme 4).
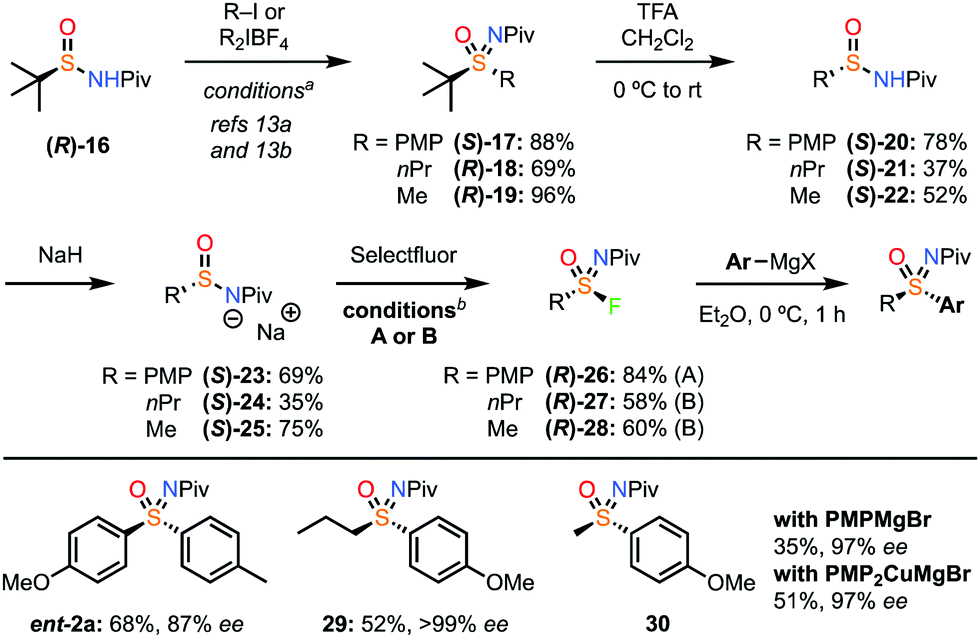 | ||
Scheme 4 Generation and reaction of enantioenriched sulfonimidoyl fluorides. a For alkylation: R–I, NaH, 15-crown-5, dioxane, 70 °C, 24 h. For arylation: R2IBF4, Cu(OTf)2 (10 mol%), iPr2EtN, DMSO, 60 °C, 24 h. b Conditions A for 23: selectfluor (2.0 equiv.), KOAc (2.0 equiv.), EtOH, 0 °C to rt, 24 h. Conditions B for 24 and 25: selectfluor (2.0 equiv.), DMF/EtOH (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2), 0 °C to rt, 24 h. :2), 0 °C to rt, 24 h. | ||
Starting from sulfinamide (R)-16, we employed the NPiv group as described by Maruoka, which was shown to be suitable for retaining ee (2a-Piv, Scheme 1) and can also be readily removed to generate the NH sulfoximine.13,25 Applying Maruoka's conditions for arylation and alkylation generated enantioenriched sulfoximines 17–19 and sulfinamides 20–22 in a process demonstrated to retain ee.13 Deprotonation gave salts 23–25. Applying the fluorination and Grignard sequence with PMP derivative (S)-23 gave high ee for sulfoximine ent-2a. On the other hand, for the alkyl derivatives an alternative set of conditions were required to ensure high conversion in the formation of the sulfonimidoyl fluorides. A mixture of DMF and EtOH was necessary to ensure both reactivity and retention of ee. We were delighted to find that the propyl and even methyl derivatives gave complete retention of ee through this fluorination and substitution process (29,30), demonstrating that racemisation is prevented, and substitution occurs without deprotonation/elimination from the alkyl sulfonimidoyl fluorides. Interestingly, deprotonation of the sulfoximine product was detected with the methyl derivative under these conditions, resulting in intermolecular attack at the Piv group.23 Instead, the use of the cuprate reagent prevented this, improved the yield of 30 and gave very high ee.
In summary, we report the preparation of enantioenriched sulfoximines using enantioenriched sulfonimidoyl fluorides. It is notable that N-Boc sulfonimidoyl fluorides react with Grignard reagents exclusively at sulfur without reduction and react stereospecifically with inversion. We report the first example of enantioenriched methyl sulfonimidoyl fluorides, and the stereospecific reaction of these motifs, avoiding elimination or racemisation. New conditions for enantiospecific fluorination of alkyl sulfinamides are presented to maximise conversion and retain ee in the sulfonimidoyl fluorides. While the NBoc and NPiv derivatives react stereospecifically, other N-groups such as Cbz and methyl carbamate are susceptible to racemisation. We expect the methods disclosed will provide further opportunities to exploit enantioenriched sulfonimidoyl fluorides and sulfoximines, particularly alkyl and methyl derivatives.
We gratefully acknowledge The Royal Society (UF140161 and URF\R\201019 to JAB), and EPSRC (DTP studentships SG and OS). We thank Dr Edward Briggs and Dr Fahima Idiris for early studies, and Dr Ulrich Lücking for valuable discussions.
Conflicts of interest
There are no conflicts to declare.References
- (a) P. Mäder and L. Kattner, J. Med. Chem., 2020, 63, 14243–14275 CrossRef PubMed; (b) Y. Han, K. Xing, J. Zhang, T. Tong, Y. Shi, H. Cao, H. Yu, Y. Zhang, D. Liu and L. Zhao, Eur. J. Med. Chem., 2020, 209, 112885 CrossRef PubMed; (c) U. Lücking, Angew. Chem., Int. Ed., 2013, 52, 9399–9408 CrossRef PubMed.
- (a) J. A. Sirvent and U. Lücking, ChemMedChem, 2017, 12, 487–501 CrossRef CAS PubMed; (b) M. Frings, C. Bolm, A. Blum and C. Gnamm, Eur. J. Med. Chem., 2017, 126, 225–245 CrossRef CAS PubMed; (c) Also: S. Wiezorek, P. Lamers and C. Bolm, Chem. Soc. Rev., 2019, 48, 5408–5423 RSC.
- (a) U. Lücking, R. Jautelat, M. Krüger, T. Brumby, P. Lienau, M. Schäfer, H. Briem, J. Schulze, A. Hillisch, A. Reichel, A. M. Wenger and G. Siemeister, ChemMedChem, 2013, 8, 1067–1085 CrossRef PubMed; (b) ClinicalTrials.gov, https://clinicaltrials.gov/ct2/show/NCT02522910 (accessed Jan 3, 2019).
- Ceralasertib from AstraZeneca (a) K. M. Foote, J. W. M. Nissink, T. McGuire, P. Turner, S. Guichard, J. W. T. Yates, A. Lau, K. Blades, D. Heathcote, R. Odedra, G. Wilkinson, Z. Wilson, C. M. Wood and P. J. Jewsbury, J. Med. Chem., 2018, 61, 9889–9907 CrossRef CAS PubMed; (b) A. Min, S.-A. Im, H. Jang, S. Kim, M. Lee, D. K. Kim, Y. Yang, H.-J. Kim, K.-H. Lee, J. W. Kim, T.-Y. Kim, D.-Y. Oh, J. Brown, A. Lau, M. J. O’Connor and Y.-J. Bang, Mol. Cancer Ther., 2017, 16, 566–577 CrossRef CAS PubMed; (c) A. G. Henssen, C. Reed, E. Jiang, H. D. Garcia, J. von Stebut, I. C. MacArthur, P. Hundsdoerfer, J. H. Kim, E. de Stanchina, Y. Kuwahara, H. Hosoi, N. J. Ganem, F. D. Cruz, A. L. Kung, J. H. Schulte, J. H. Petrini and A. Kentsis, Sci. Transl. Med., 2017, 9, eaam9078 CrossRef PubMed.
- For other bioactive compounds containing sulfoximines, see: (a) C. Gege, F. J. Bravo, N. Uhlig, T. Hagmaier, R. Schmachtenberg, J. Elis, A. Burger-Kentischer, D. Finkelmeier, K. Hamprecht, T. Grunwald, D. I. Bernstein and G. Kleymann, Sci. Transl. Med., 2021, 13, eabf8668 CrossRef CAS PubMed; (b) Also see ref. 1, and: U. Lücking, Org. Chem. Front., 2019, 6, 1319–1324 RSC.
- M. Andresini, A. Tota, L. Degennaro, J. A. Bull and R. Luisi, Chem. – Eur. J., 2021, 27, 17293–17321 CrossRef CAS PubMed.
- (a) V. Bizet, C. M. M. Hendriks and C. Bolm, Chem. Soc. Rev., 2015, 44, 3378–3390 RSC; (b) J. A. Bull, L. Degennaro and R. Luisi, Synlett, 2017, 2525–2538 CrossRef CAS.
- For metal free processes: (a) M. Zenzola, R. Doran, L. Degennaro, R. Luisi and J. A. Bull, Angew. Chem., Int. Ed., 2016, 55, 7203–7207 CrossRef CAS PubMed; (b) A. Tota, M. Zenzola, S. J. Chawner, S. S. John-Campbell, C. Carlucci, G. Romanazzi, L. Degennaro, J. A. Bull and R. Luisi, Chem. Commun., 2017, 53, 348–351 RSC; (c) S. Chaabouni, J.-F. Lohier, A.-L. Barthelemy, T. Glachet, E. Anselmi, G. Dagousset, P. Diter, B. Pégot, E. Magnier and V. Reboul, Chem. – Eur. J., 2018, 24, 17006–17010 CrossRef CAS PubMed.
- For selected nitrogen transfer with transition metal catalysis: see: (a) H. Okamura and C. Bolm, Org. Lett., 2004, 6, 1305–1307 CrossRef CAS PubMed; (b) M. Zenzola, R. Doran, R. Luisi and J. A. Bull, J. Org. Chem., 2015, 80, 6391–6399 CrossRef CAS PubMed; (c) H. Yu, Z. Li and C. Bolm, Angew. Chem., Int. Ed., 2018, 57, 324–327 CrossRef CAS PubMed.
- T. Q. Davies, M. J. Tilby, J. Ren, N. A. Parker, D. Skolc, A. Hall, F. Duarte and M. C. Willis, J. Am. Chem. Soc., 2020, 142, 15445–15453 CrossRef CAS PubMed.
- B. Gao, S. Li, P. Wu, J. E. Moses and K. B. Sharpless, Angew. Chem., Int. Ed., 2018, 57, 1939–1943 CrossRef CAS PubMed.
- Z. P. Shultz, T. Scattolin, L. Wojtas and J. M. Lopchuk, Nat. Synth., 2022, 1, 170–179 CrossRef.
- (a) Y. Aota, T. Kano and K. Maruoka, Angew. Chem., Int. Ed., 2019, 58, 17661–17665 CrossRef CAS PubMed; (b) Y. Aota, T. Kano and K. Maruoka, J. Am. Chem. Soc., 2019, 141, 19263–19268 CrossRef CAS PubMed.
- (a) M. R. Jones and D. J. Cram, J. Am. Chem. Soc., 1974, 96, 2183–2190 CrossRef CAS; (b) C. R. Johnson, E. U. Jonsson, A. Wambsgans and C. C. Bacon, J. Org. Chem., 1979, 44, 2061–2065 CrossRef CAS.
- C. R. Johnson, K. G. Bis, J. H. Cantillo, N. A. Meanwell, M. F. D. Reinhard, J. R. Zeller and G. P. Vonk, J. Org. Chem., 1983, 48, 1–3 CrossRef CAS.
- Also see: (a) M. Harmata and R. J. Claassen, Tetrahedron Lett., 1991, 32, 6497–6500 CrossRef CAS; (b) D. van Leusen and A. M. van Leusen, Recl. des Trav. Chim. des Pays-Bas, 2010, 103, 41–45 CrossRef; (c) M. Reggelin, H. Weinberger and V. Spohr, Adv. Synth. Catal., 2004, 346, 1295–1306 CrossRef CAS; (d) For TMSCF3 as a nucleophile, see: R. Kowalczyk, A. J. F. Edmunds, R. G. Hall and C. Bolm, Org. Lett., 2011, 13, 768–771 CrossRef CAS PubMed; (e) For a single example reacting MeLi with trifluoromethyl sulfonimidate, see: M. Wright, C. Martínez-Lamenca, J. E. Leenaerts, P. E. Brennan, A. A. Trabanco and D. Oehlrich, J. Org. Chem., 2018, 83, 9510–9516 CrossRef CAS PubMed.
- (a) M. Reggelin and B. Junker, Chem. – Eur. J., 2001, 7, 1232–1239 CrossRef CAS PubMed; (b) P. Mendonça Matos, W. Lewis, S. P. Argent, J. C. Moore and R. A. Stockman, Org. Lett., 2020, 22, 2776–2780 CrossRef PubMed; (c) P. M. Matos and R. A. Stockman, Org. Biomol. Chem., 2020, 18, 6429–6442 RSC; (d) P. M. Matos, W. Lewis, J. C. Moore and R. A. Stockman, Org. Lett., 2018, 20, 3674–3677 CrossRef CAS PubMed.
- S. Greed, E. L. Briggs, F. I. M. Idiris, A. J. P. White, U. Lücking and J. A. Bull, Chem. – Eur. J., 2020, 26, 12533–12538 CrossRef CAS PubMed.
- D. Liang, D. E. Streefkerk, D. Jordaan, J. Wagemakers, J. Baggerman and H. Zuilhof, Angew. Chem., Int. Ed., 2020, 59, 7494–7500 CrossRef CAS PubMed.
- H. Mukherjee, J. Debreczeni, J. Breed, S. Tentarelli, B. Aquila, J. E. Dowling, A. Whitty and N. P. Grimster, Org. Biomol. Chem., 2017, 15, 9685–9695 RSC.
- D. D. Liang, S. P. Pujari, M. Subramaniam, M. Besten and H. Zuilhof, Angew. Chem., Int. Ed., 2022, 61, e202116158 CAS.
- This hypothesis was supported by reports of degenerate exchange on fluorosulfates and sulfamoyl fluorides. (a) Q. Zheng, H. Xu, H. Wang, W. G. H. Du, N. Wang, H. Xiong, Y. Gu, L. Noodleman, K. B. Sharpless, G. Yang and P. Wu, J. Am. Chem. Soc., 2021, 143, 3753–3763 CrossRef CAS PubMed; (b) M. H. Jeon, Y. Do Kwon, M. P. Kim, G. B. Torres, J. K. Seo, J. Son, Y. H. Ryu, S. Y. Hong and J. H. Chun, Org. Lett., 2021, 23, 2766–2771 CrossRef PubMed.
- See ESI† for further details.
- Grignard reagents were used as supplied or prepared by halogen exchange using iPrMgCl·LiCl, with comparable results.
- Alkylation of NBoc sulfinamides were successful, but cleavage of the NBoc group occurred preferentially to cleavage of the S–tBu bond on treatment with TFA.
Footnote |
| † Electronic supplementary information (ESI) available: Further optimisation including for organocuprate and organozinc reagents, determination of the stereochemical outcome, reactions using stabilised lithium anions, synthesis of enantioenriched sulfinamide salts, experimental procedures, characterisation data, HPLC data. See DOI: https://doi.org/10.1039/d2cc01219g |
| This journal is © The Royal Society of Chemistry 2022 |