
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Selective coordination of coinage metals using orthogonal ligand scaffolds
Vanitha R.
Naina†
,
Frederic
Krätschmer†
and
Peter W.
Roesky
 *
*
Institute of Inorganic Chemistry, Karlsruhe Institute of Technology, Engesserstr. 15, 76131 Karlsruhe, Germany. E-mail: roesky@kit.edu
First published on 13th April 2022
Abstract
Group 11 metal complexes with their ability to form metallophilic interations are widely pursued to develop multifunctional luminescent materials. Heteronuclear coinage metal complexes are promising candidates to tune electronic and optical properties which are not readily accessed by their homometallic congeners. In this review, we present the concept of orthogonal ligands which are rationally designed to access heteronuclear coinage metal complexes and studied in terms of their photophysical properties. Bifunctional ligands containing soft and hard donor atoms have the potential of providing different coordination modes to selectively synthesise heterobimetallic complexes in a predictable manner. This review deals with ligand sets composed of pyridine, bipyridine- or iminopyridine-substituted NHCs featuring C–N coordination modes, phosphine-based N-heterocycles and amidinate ligand scaffolds comprising of P–N functionalities and mixed phosphine–phosphine oxide with P–O donor sites. Therefore, the scope of this perspective is the discussion of heteronuclear coinage metal complexes supported by recently developed bifunctional ligands in terms of their synthesis, coordination geometries and tunability of optical properties when compared to their homometallic analogues.
Introduction
Light harvesting devices such as OLEDs or solar cells have attracted great attention as they are promising means to harness renewable energy sources (solar energy).1 Coinage metal complexes have found potential applications in the field of organic photovoltaics owing to their remarkable optical properties. Group 11 metals in +1 oxidation state are known for their ability to form d10–d10 interactions termed as “metallophilicity”.1–6 For gold, these “aurophilic interactions”4,5 arise only when the distance between two metal atoms falls below 3.5 Å i.e. less than the sum of the van der Waals radii of two gold atoms. Although less known, these interactions were also observed for coinage congeners (Cu and Ag) with closed shell electronic configuration. They are referred as “cuprophilic” with distance between Cu(I) cations below 2.8 Å7,8 and “argentophilic” interactions when the distance between two silver(I) cations is in the range of 2.9–3.4 Å.6,9,10 In principle, these bonds are supported by a number of ligands, hence, can be modulated by fine tuning of the ligand backbone,11 solvents12etc. These interactions not only play a crucial role in stabilising supramolecular structures but also in determining the optical properties of the metal complexes.13–16 A critical review on homometallic and heterometallic clusters involving these weak d10–d10 attractive interactions was published by Braunstein and co-workers in 2011.17A wealth of literature is available on coinage metal complexes with interesting properties such as thermochromism, vapochromism, etc.12,18–23 Structurally well-characterised heterometallic assemblies have been widely investigated in order to establish structure–property correlations and to pursue multifunctional light-emitting materials. The presence of different metal ions in a molecular architecture often leads to distinct properties (due to synergistic effects) when compared to their analogues of monometallic combinations.13,24 This review is intended to be of tutorial nature wherein, we particularly focus on a selective set of rationally designed orthogonal ligands which allow the synthesis of multimetallic complexes, in which the metals are arranged in defined compartments showing supported metallophilic interactions in many cases. These compounds were systematically studied with respect to their photoluminescence properties.
Multiple comprehensive reviews focusing on the properties of polynuclear coinage metal complexes have been reported.25–27 Recently, Koshevoy, Grachova and co-workers published a review devoted to optical properties of multinuclear coinage metal complexes based on bridging phosphine ligands.28 Even though, phosphines have affinity for all the group 11 metals with d10-electronic configuration and often result in fascinating properties,29,30 it is difficult to selectively design heterometallic complexes due to the scrambling nature of metal ions between different phosphorous donor sites in solution state.31 Hence, it is of no doubt that the synthesis of stable heterometallic complexes demand a rational ligand engineering, which can selectively coordinate to different metal ions. In group 11 chemistry, the formation of heterometallic complexes has been used for the synthesis of luminescent materials. However, the challenge is the selective assembly of such compounds.
Orthogonal ligands, commonly referred as bifunctional ligands, can be defined as organic ligands containing two different coordinating sites, which can selectively coordinate to two different metal ions. From conventional Pearson concept of hard and soft acid and bases (HSAB),32,33 in group 11 Au(I) prefers coordinating to soft donors (e.g., P) in comparison to hard donors (e.g., N, O), whereas Cu(I) has a greater affinity for the hard donor atoms and Ag(I) is somewhere in between. Following this principle, ligands having several heteroatoms as coordination sites are suitable for the selective construction of heterometallic architectures in order to tune the properties of interest. Moreover, multidentate ligands allow several combinations (Fig. 1) and spatial proximity of different metal ions leading to metal–metal interactions. This point of view has led to the development of ligands with tailor-made steric and electronic properties. Earliest example of orthogonal ligand based heteronuclear complex was reported by Che and co-workers in 1998.34 Having emphasised the principal role of multidentate ligands with heteroatoms, it is noteworthy that there are other ways to achieve the focused goal e.g., introducing coordinating anions like CN-,35,36 triflate,37 or solvents such as acetonitrile and pyridine.38–40
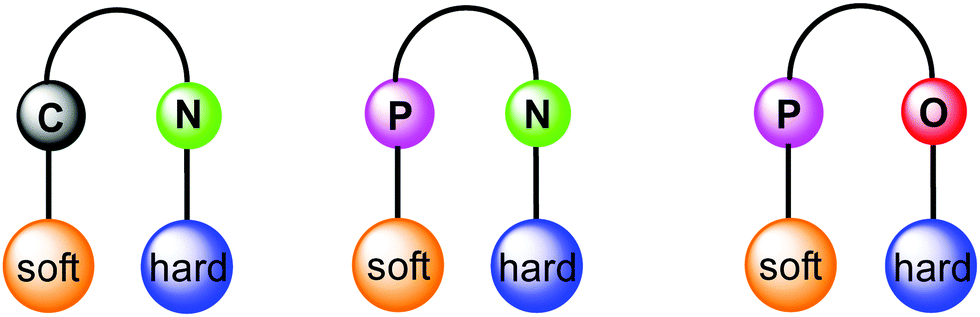 | ||
| Fig. 1 Schematic representation of orthogonal ligand scaffolds with selective coordination to metal ions following the HSAB principle. | ||
Herein, we particularly intend to discuss a limited set of ligands, which are reported in the past few years. This review has been categorised into three sub-groups based on the coordination modes present in the ligand namely C–N, P–N, P–O.
C–N coordination
Coordination modes
Complexes of the coinage metals with N- and C-donor ligands are embossed by tetrahedral, trigonal planar and linear coordination modes. To investigate on this behaviour, a series of ligands with rather soft carbene donor functionalities for gold(I) atoms and a tethered pyridine, bipyridine or imino-pyridine moiety as hard donor for copper(I) and silver(I) atoms (L1–L6) were used (Schemes 1, 2, 4, 6, and 7).20,22,41–46 In case of gold(I), linear coordination with soft carbene donor functionalities was obtained for all compounds shown in Schemes 1, 2, 4, 6, and 7.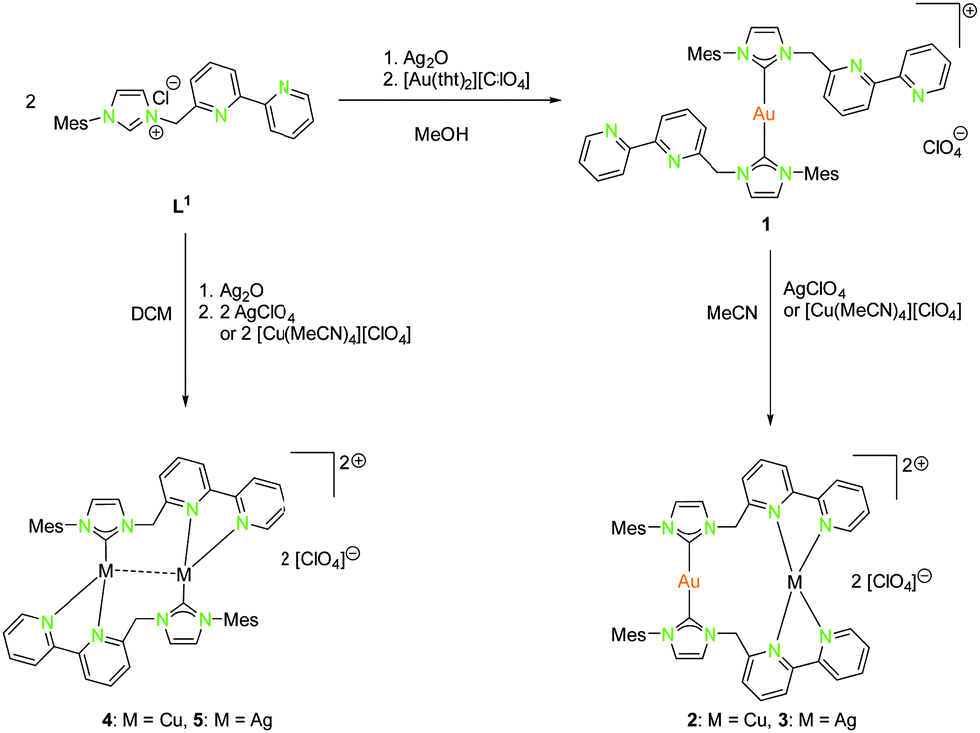 | ||
| Scheme 1 Synthesis of mononuclear (1), dinuclear homo- (4, 5) and heterobimetallic (2, 3) compounds with NHCbipy (L1).41 | ||
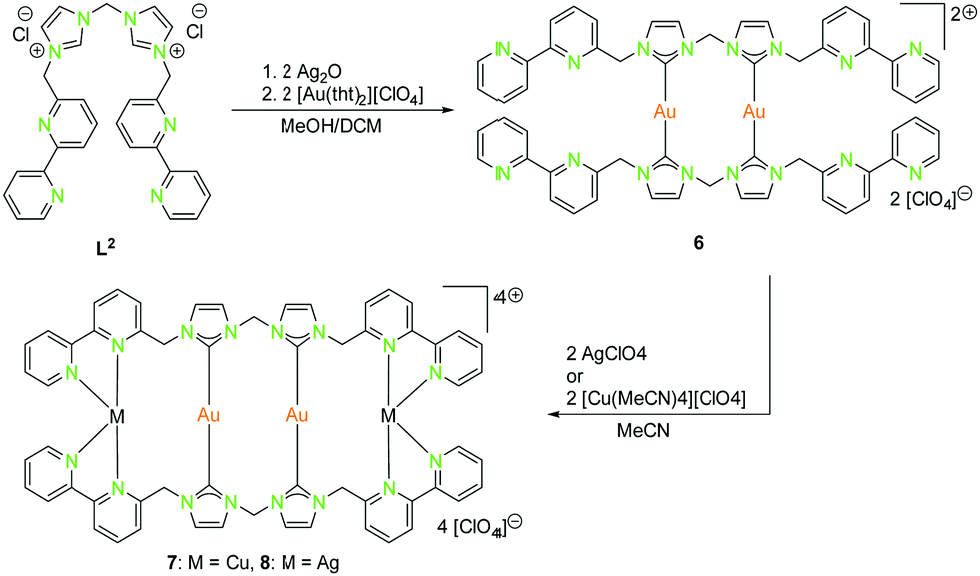 | ||
| Scheme 2 Synthesis of homometallic dinuclear (6) and heterometallic tetranuclear (7, 8) bis-NHCbipy (L2) compounds.42 | ||
 | ||
| Scheme 3 V-shaped (left) and zig-ag chain (right) structures of compound 8.42 | ||
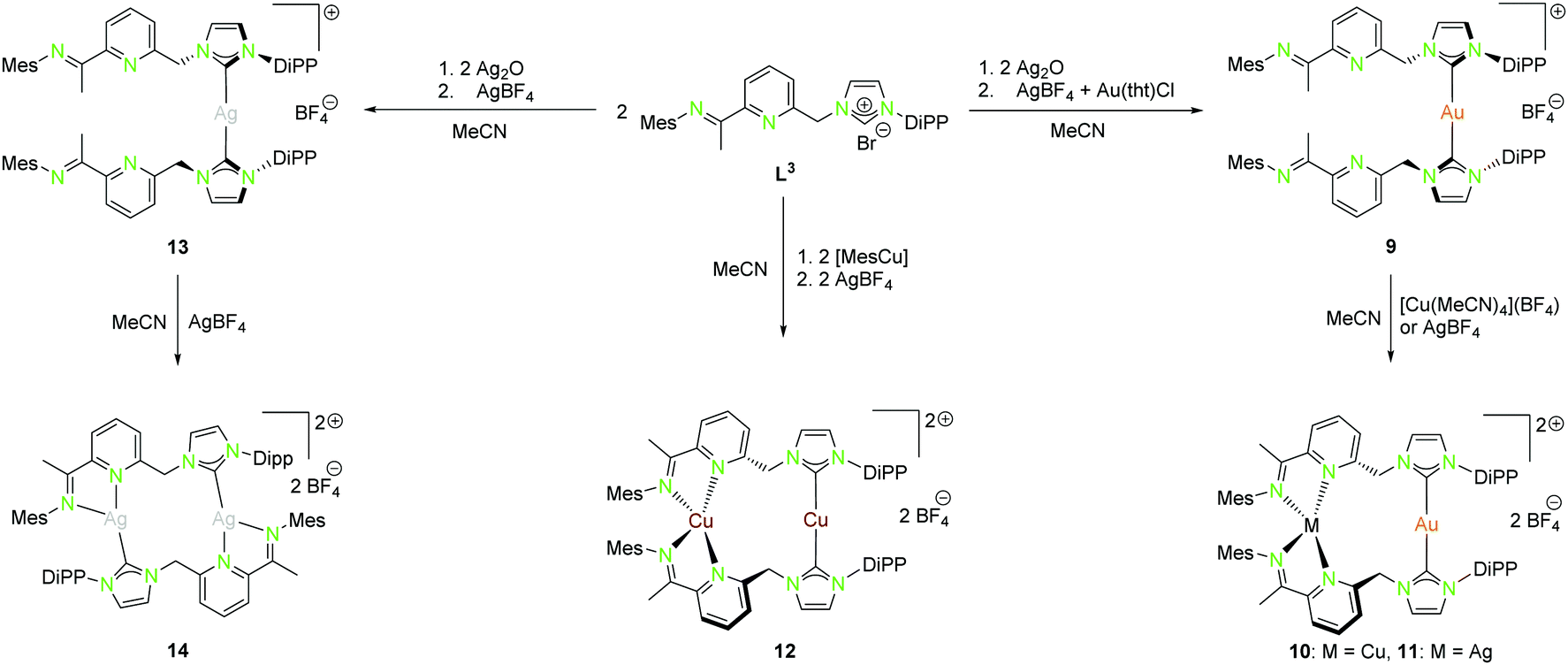 | ||
| Scheme 4 Synthesis of mononuclear (9, 13), as well as homo- (12, 14) and heterobimetallic (10, 11) NHCimpy (L3) compounds.43 | ||
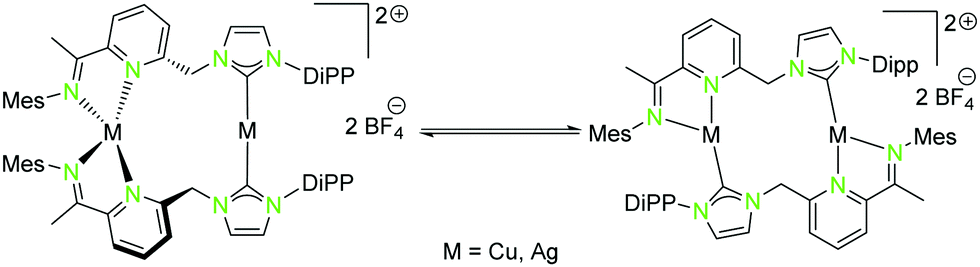 | ||
| Scheme 5 Isomerisation of homometallic NHCimpy (L3) compounds (12, 14) in solution.43 | ||
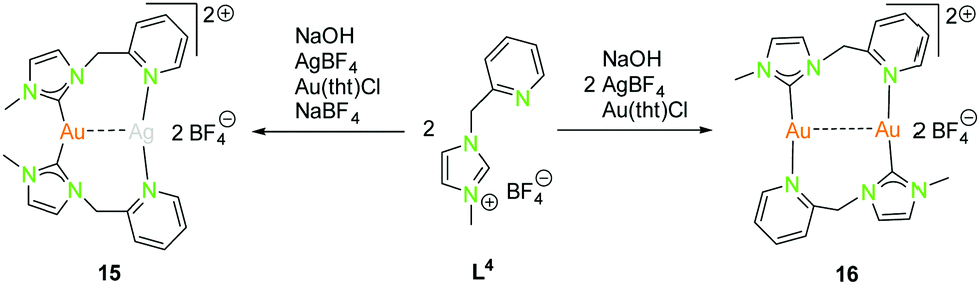 | ||
| Scheme 6 Synthesis of heterometallic dinuclear (15) and homometallic dinuclear (16) NHCpy (L4) compounds.47 | ||
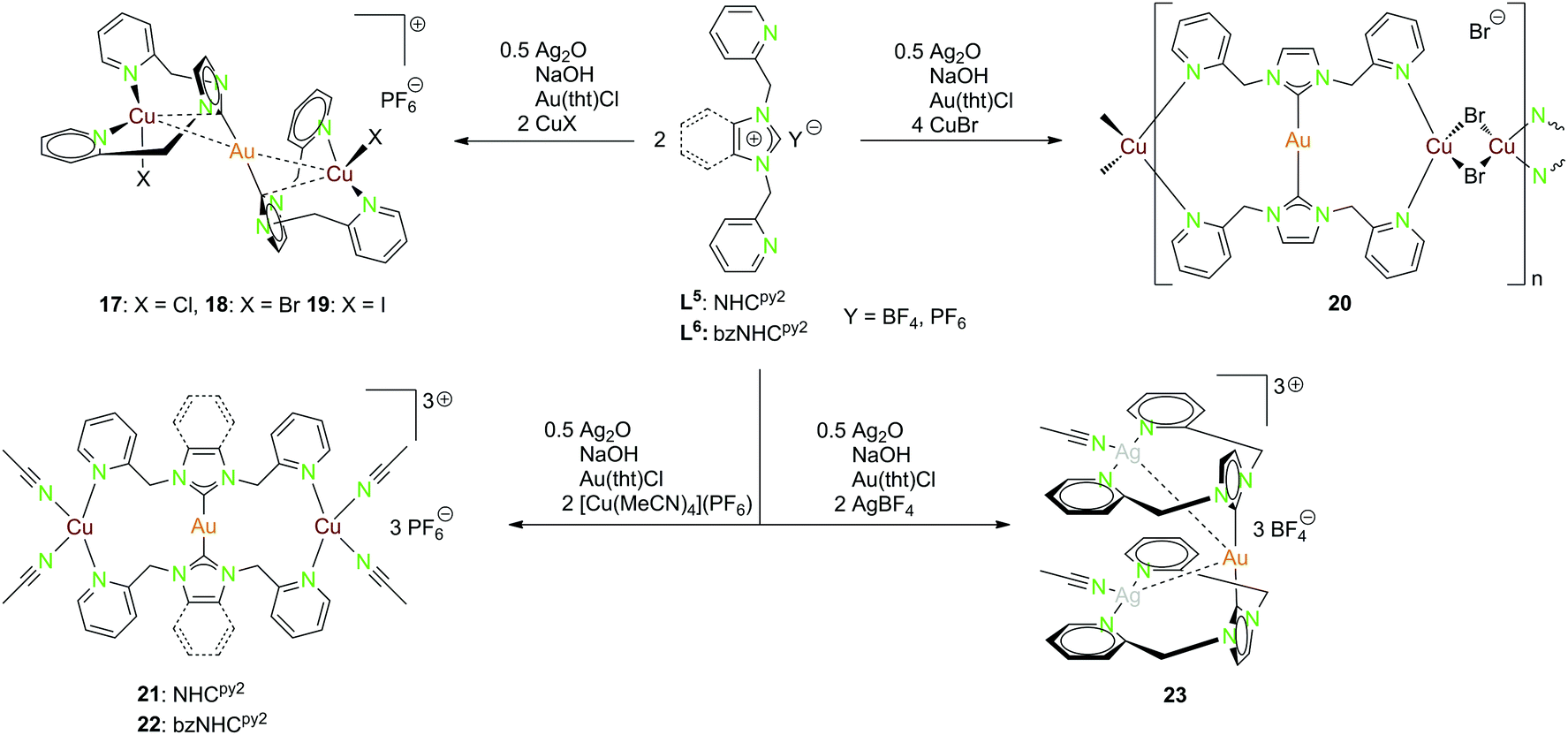 | ||
| Scheme 7 Synthesis of the heterotrinuclear complexes 17–23 with NHCpy2 (L5) and bzNHCpy2 (L6).20,22,45,46 | ||
By using Ag2O for deprotonation of L1 followed by transmetallation with a gold(I) salt, the monometallic gold(I) compound 1 was synthesised (Scheme 1). Treatment of 1 with silver(I) or copper(I) salts resulted in the formation of heterobimetallic compounds 2 and 3 (Scheme 1 (right)).41 The coordination of gold(I) to the bipyridine moiety was not observed, as expected for a soft metal. Starting with L1, the homobimetallic silver(I) complex 4 was obtained from the corresponding silver(I) precursors, while compound 5 was synthesised by transmetallation of an in situ generated silver(I) species with a copper(I) salt (Scheme 1 (left)).41 Copper(I) and silver(I) prefer the hard N-donor functionalities. Nevertheless, in the absence of gold(I) also coordination to soft donors concomitant with the formation of homometallic complexes is possible. Copper(I) is most often tetrahedrally coordinated but can also form trigonal planar or linear arrangements (Schemes 1, 2, 4, 5, and 7).
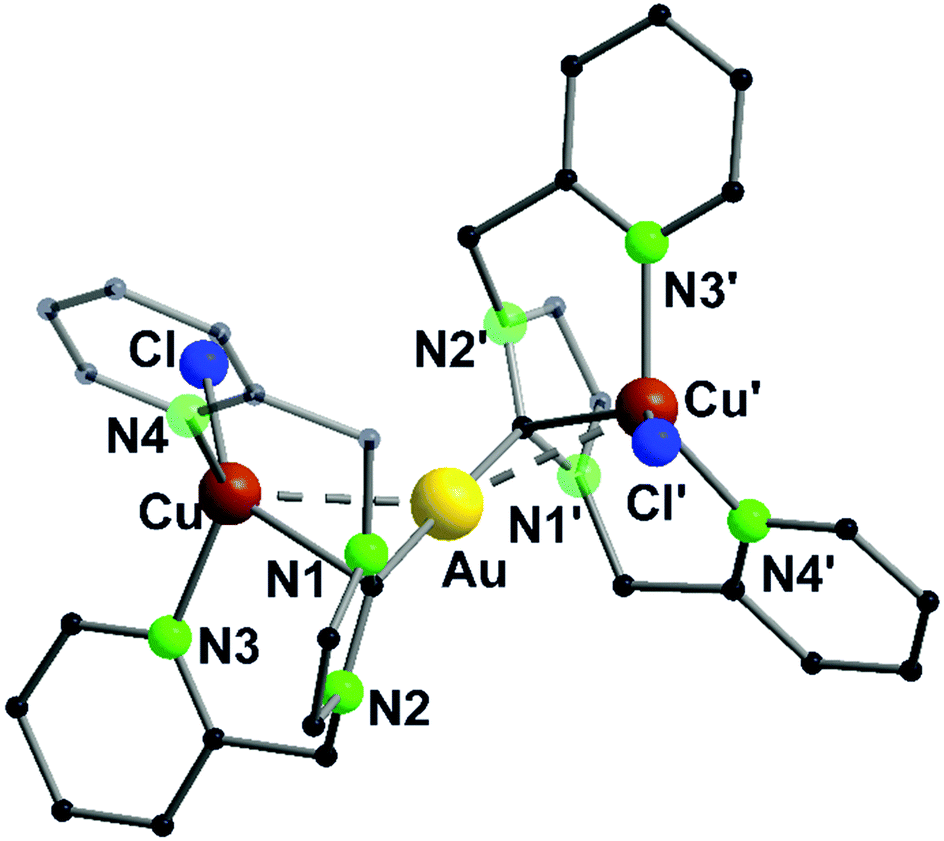
The propensity of silver(I) to adopt low coordination numbers and a linear coordination is reflected by the almost linear arrangement of N–Ag–N′ (> 160°) in the bipyridine units (3). In addition, there is a higher tendency of silver(I) to form metallophilic contacts, which is reflected by the smaller intermetallic distance Ag–Ag (3.1034(11) Å) (5) compared to the copper(I) homologue (3.2168(9) Å) (4) despite the larger van der Waals radius (Scheme 1 and Fig. 2).41
 | ||
| Fig. 2 Molecular structure of 5 in the solid state. Hydrogen atoms and the counter ions are omitted for clarity.41 | ||
With the intention of synthesising tetranuclear coinage metal chains the bis-NHCbipy ligand L2, composed out of two L1 moieties, was designed.42 The synthesis of the coinage metal complexes 6–8 (Scheme 2) was achieved in a similar fashion to compounds 1–3.41 While the homobimetallic gold(I) compound 6 does not show intermetallic interactions, the coordination of copper(I) atoms into the bipyridine moieties in compound 7 leads to a V-shaped structure with a shorter Au–Au distance of 3.38 Å and Au–Cu distances of around 4.95 Å (Scheme 3). In contrast silver(I) complex 8, which is ligated by L2, shows two molecules in the asymmetric unit, one homologue to the V-shaped copper(I) compound 7 without metallophilic interactions (Au–Au 3.80 Å, Au–Ag ca. 5.16 Å), and a zig-zag chain structure with metallophilic interactions between all neighbouring atoms (Au–Au 3.05 Å, Au–Ag ca. 2.97 Å) (Scheme 3). This structural arrangement can only be achieved by a folding of the ligand.42
A rather similar ligand to L1 is the NHCimpy system L3, in which the bipyridine moiety was exchanged by an iminopyridin scaffold (Scheme 4).43 The synthesis of the coinage metal complexes ligated to L3 is shown in Scheme 4. They were obtained in a similar synthetic approach as used for the synthesis of the compounds discussed above. Only the homobimetallic copper(I) compound 12 was synthesised differently by deprotonation of L3 with mesityl copper(I) followed by halide abstraction with AgBF4.43
The solid state structure of the homometallic silver(I) imino-pyridine complex 14 shows a distorted trigonal planar coordination (“head to tail”), whereas for the copper(I) compound 12 a linear and a tetrahedral distorted coordination (“head to head”) was found. Nevertheless, these two compounds isomerise in solution and thus form both coordination modes (Scheme 5).
The heterobimetallic compounds 10 and 11 show “head to head” arrangement, which is in contrast to the homobimetallic silver(I) complex 14 probably due to the formation of stronger Au–NHC bonds and the aversion of gold(I) to hard donor sites.43 Like the homobimetallic compounds 12 and 14, the heterobimetallic compounds show no metallophilic interactions with intermetallic distances largely exceeding the van der Waals radii (Au–Cu 4.91 Å, Au–Ag 5.14 Å).43
The following compounds from Catalano et al. are quite similar NHC ligands, but instead of an attached bipyridine functionality, which is favourable for tetrahedral coordination, pyridine units are used.20,22,45–47 This results in different coordination modes and leaves space for coordinating solvent molecules.
The dinuclear NHCpy (L4) complexes 15 and 16 can be synthesised by deprotonation through NaOH, addition of AgBF4 leads to [(NHCpy)2Ag][BF4] and is transferred to [(NHCpy)2Au][BF4] using Au(tht)Cl, followed by the addition of AgBF4 or in situ synthesised Au(tht)BF4 (Scheme 6).47
The heterometallic gold(I)–silver(I) complex 15 is arranged in a “head to head” fashion, where gold(I) is coordinated in a distorted linear geometry by two NHC's and silver(I) is coordinated to two pyridyl moieties, not considering the metallophilic interaction (Au–Ag 3.03 Å).47 The C–Au–C angle is 170.6°, which is more linear than the N–Ag–N angle of 154.6°. The homometallic gold(I) compound 16 is in a “head to tail” fashion with gold(I) coordinated to one NHC and a pyridyl moiety in a nearly linear manner.47 The “head to head” coordination is inconvenient due to the fact, that one gold atom would be coordinated by two hard nitrogen donors which is in contradiction to the HSAB principle. The bond angles C–Au–N are close to 180° (179.4° and 178.5°). The intermetallic distance (Au–Au 3.17 Å) is slightly longer than in the heteronuclear compound 15 (Au–Ag 3.03 Å).47
Multiple heterometallic gold(I)–copper(I) halide compounds (17–20) were synthesised using the NHCpy2 ligand (L5).46 The ligand was deprotonated by NaOH and Ag2O, with transmetallation to the [(NHCpy2)2Au](PF6) complex using Au(tht)Cl and followed by addition of copper(I)–halides (Scheme 7).46
Compounds 17–19 are isostructural, with gold(I) being nearly linear coordinated by the two ligand moieties. Each copper(I) is coordinated by two pyridyl functionalities of the same ligand, as well as one halide and a short contact to the NHC carbon. The gold(I)–copper(I) separations range from 2.6688(9) Å in 18 over 2.6786(10) Å in 19 to 2.7030(5) Å in 17 (Fig. 3).46
 | ||
| Fig. 3 Molecular structure of 17 in the solid state. Hydrogen atoms and the counter ions are omitted for clarity.46 | ||
Compound 20 forming a polymeric structure shows an almost linear coordinated gold(I) centre but unlike the previous compounds the two copper(I) centers coordinate to two pyridyl units at opposed ligands. The distorted tetrahedral coordination sphere is completed by two bridging bromide ions. No intermetallic interactions occur for this compound.46 The trinuclear heterometallic compounds 21 and 22 can be isolated when reacting the ligands NHCpy2 (L5) and bzNHCpy2 (L6) with [Cu(MeCN)4](PF6) instead of copper(I) halides (Scheme 7) (Fig. 3).20,22
Gold(I) is nearly linear coordinated by two carbon atoms, while the two copper(I) centers are coordinated by two pyridyl functionalities and two acetonitrile molecules in a distorted tetrahedral geometry. The Au–Cu separation is in both cases around 4.6 Å, the replacement of acetonitrile by one methanol (21) or the complete abstraction (22) leads to considerably shorter intermetallic contacts (2.8 Å, 3.0 Å).20,22
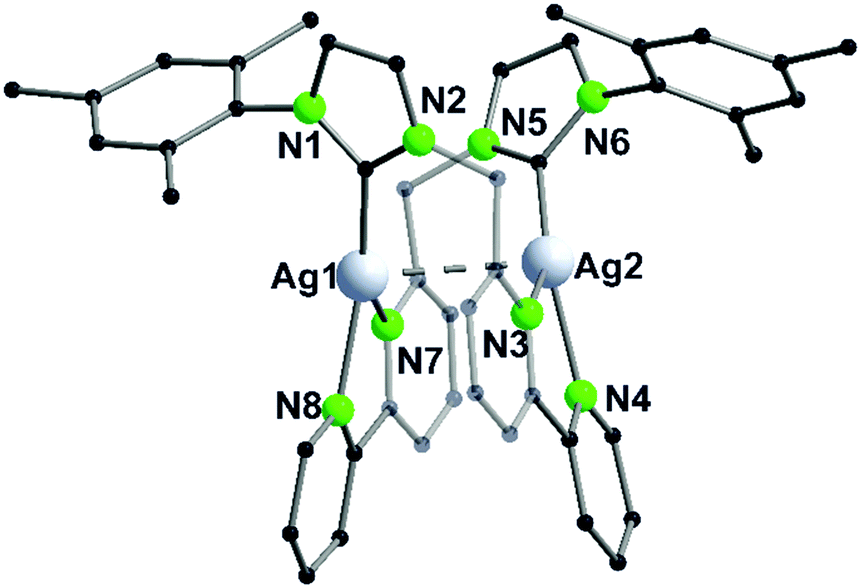
The gold(I)–silver(I) trinuclear compound 23 was synthesised using NHCpy2 (L5), following the pathway mentioned above, with the addition of two equivalents AgBF4 in the last step (Scheme 7).45 The gold(I) centre is coordinated by the two NHC carbon atoms. Like in compound 17–19, silver(I) is coordinated by the pyridyl units of the same ligand and one acetonitrile molecule in a T-shaped geometry. Therefore, the pyridine rings are splayed back, achieving an almost orthogonal position to the imidazole ring.45 The intermetallic distances between gold(I) and silver(I) are about 3.25 Å and the silver(I)–silver(I) distance is 3.43 Å close to argentophilic interactions (Fig. 4).45
 | ||
| Fig. 4 Molecular structure of 23 in the solid state. Hydrogen atoms and the counter ions are omitted for clarity.45 | ||
All mentioned complexes follow the HSAB principle, showing the coordination of gold(I) with low coordination numbers and almost exclusively soft donor sites. Copper(I) mostly shows tetrahedral coordination or in rare cases a coordination between linear and tetrahedral arrangement always looking for high coordination numbers with hard donors. In the case of silver(I), it can be emphasised that it lies in between, wanting low coordination numbers but coordinates to hard as well as soft donors.
Photoluminescence properties
For compounds 1–14 shown above (Schemes 1, 2 and 4), a vibronically structured greenish PL emission around 480 nm is observed. The exceptions are the mononuclear gold(I) compound 9, the homobimetallic copper(I) complex 4 and the gold(I)–copper(I) heterobimetallic complexes (2, 7 and 10), where the presence of the copper(I) leads to a broad band at higher wavelengths. This already indicates a strong influence of the copper(I) ion in heterobimetallic complexes to their PL properties. The spectra of compounds 1–8, which contain one of the NHCbipy ligands (L1 or L2) resemble each other (Fig. 5 and 6),41,42 since ligand L2 is formally built from two of the coordinating units of L1. The vibronic structures in the spectra of these compounds were assigned to intraligand (IL) transition centered on the bipyridine moieties.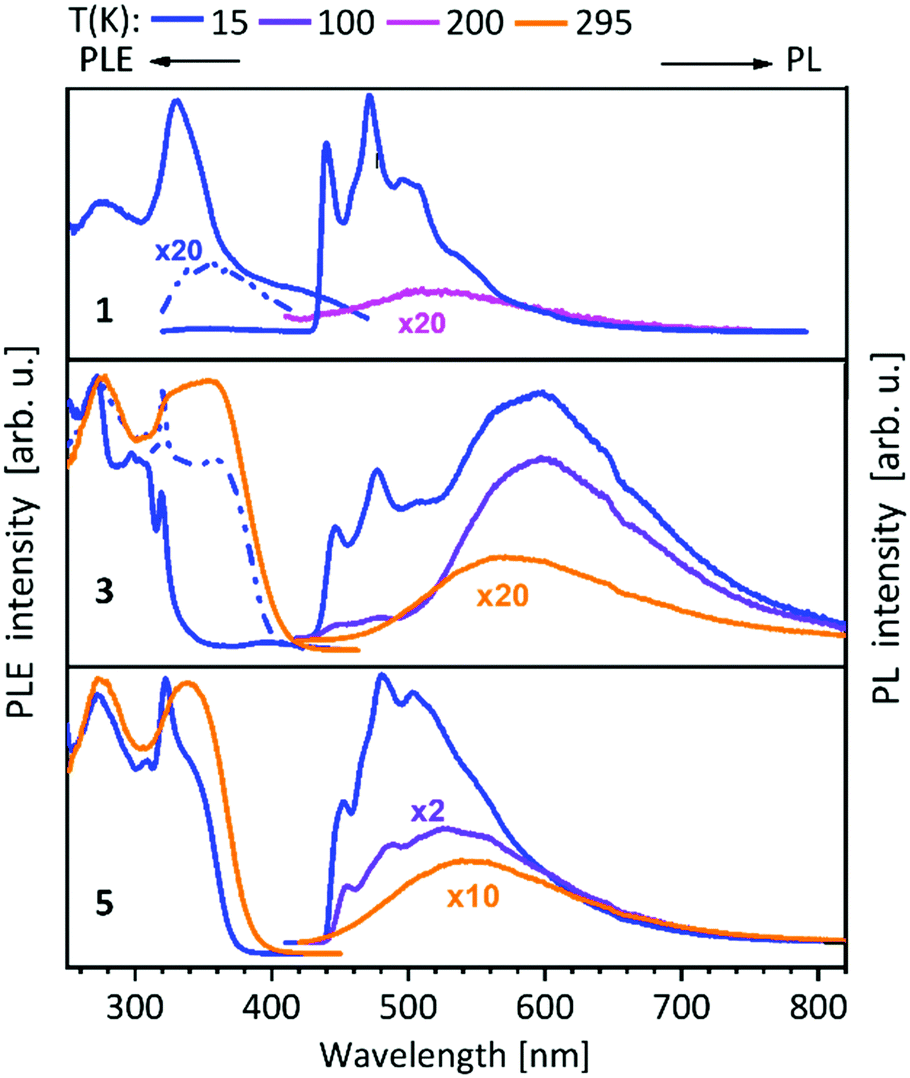 | ||
| Fig. 5 Solid state excitation and emission spectra of NHCbipy (L1) metal complexes 1, 3, 5. PL was excited at λexc = 300 nm and PLE spectra were recorded at λem = 540/540 nm (1), λem = 480; 580/580 nm (3) and λem = 550/550 nm (5).41 | ||
 | ||
| Fig. 6 Solid state excitation and emission spectra of bis-NHCbipy (L2) metal complexes 6–8. PL was excited at λexc = 300 nm for 6, 8 and λexc = 320 nm for 7. PLE spectra were recorded at λem = 500/560 nm (6), λem = 700/750 nm (7) and λem = 550/600 nm (8) at low/elevated temperatures respectively.42 | ||
The PLE onset at around 400–440 nm is consistent with the colourless and yellowish colour of the compounds, but in discrepancy with the orange to brown copper(I) containing compounds. The contributions from the d-orbitals of copper(I) lead to a significant decrease of the HOMO–LUMO gap, resulting in the redshift of absorption.41,42 This was explained by TDDFT calculations by using the trinuclear di(bipyridine)phenylphosphine ligated Cu–Au–Cu complex 32 as model compound (see below and Scheme 10).48 Also, the participation of copper(I) with photoexcitation into lower-energy excited states results in very efficient non-radiative relaxation. In contrast, the contribution by silver(I) and gold(I) atoms occurs at comparably low energies, thus these orbitals do not play a role for the excitations and have no significant influence on the optical properties.49 The PL spectra of the L3 ligated complexes (9–14) (Fig. 7) resemble the ones discussed above (compounds 1–8) (Fig. 5 and 6).41,43
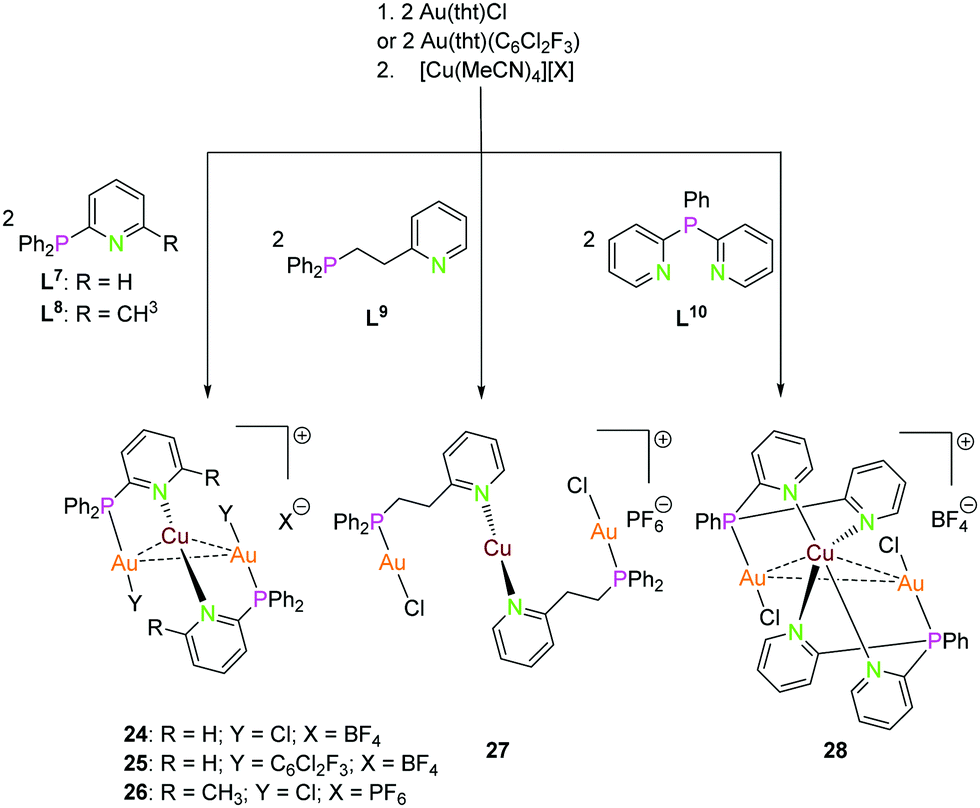 | ||
| Scheme 8 Synthesis of trinuclear heterometallic Ppy (L7–L10) compounds 24–28.13,51,54 | ||
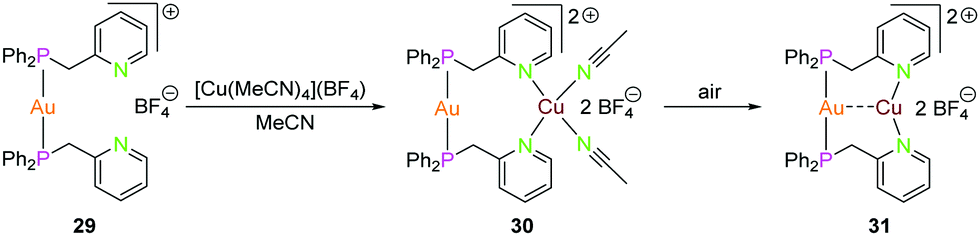 | ||
| Scheme 9 Synthesis of dinuclear heterometallic compounds 29–31 with Ppy ligand (L11).55 | ||
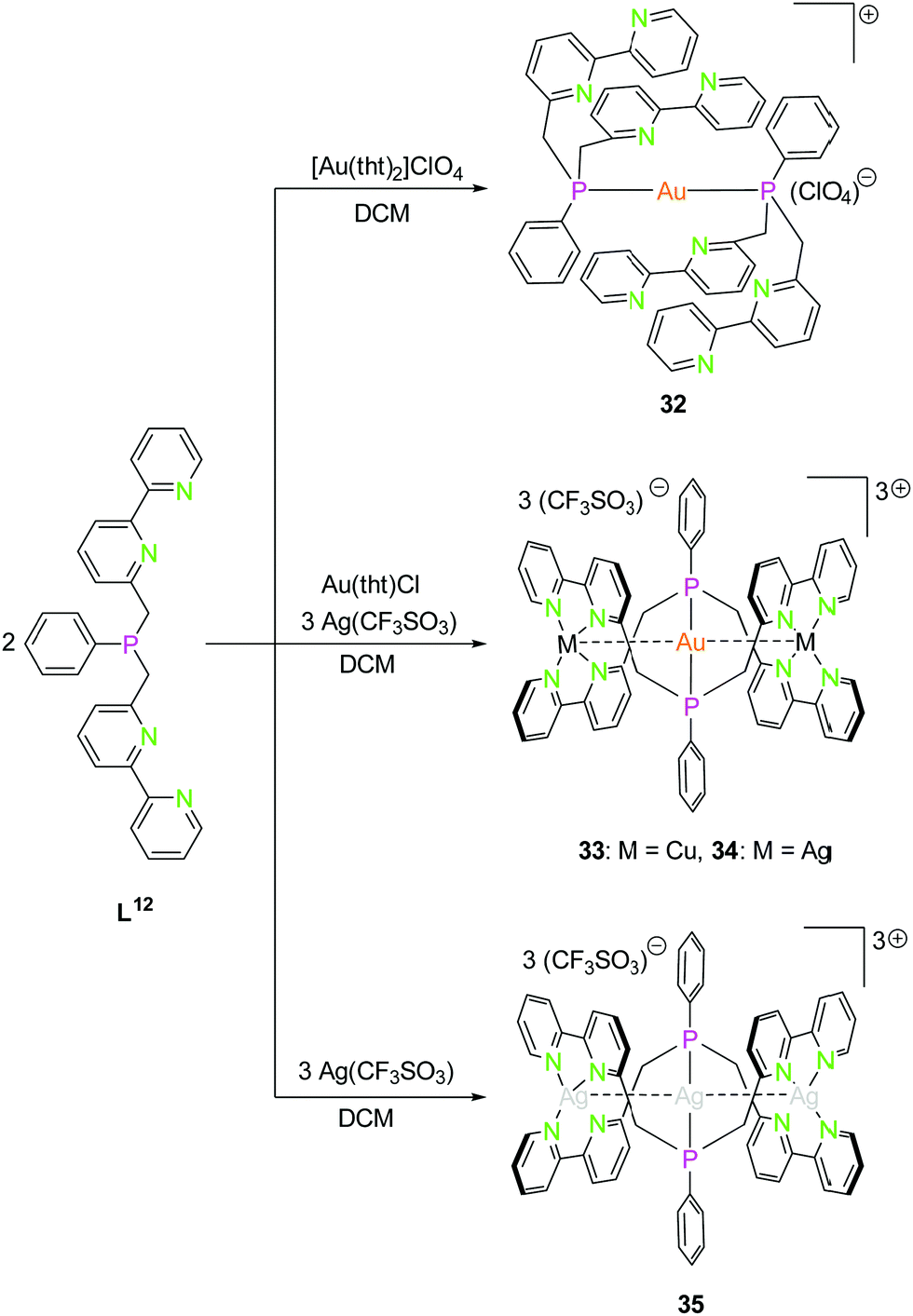 | ||
| Scheme 10 Synthesis of mononuclear gold(I) (32) and trinuclear hetero- (33, 34) and homometallic silver(I) (35) PN4 (L12) compounds.48 | ||
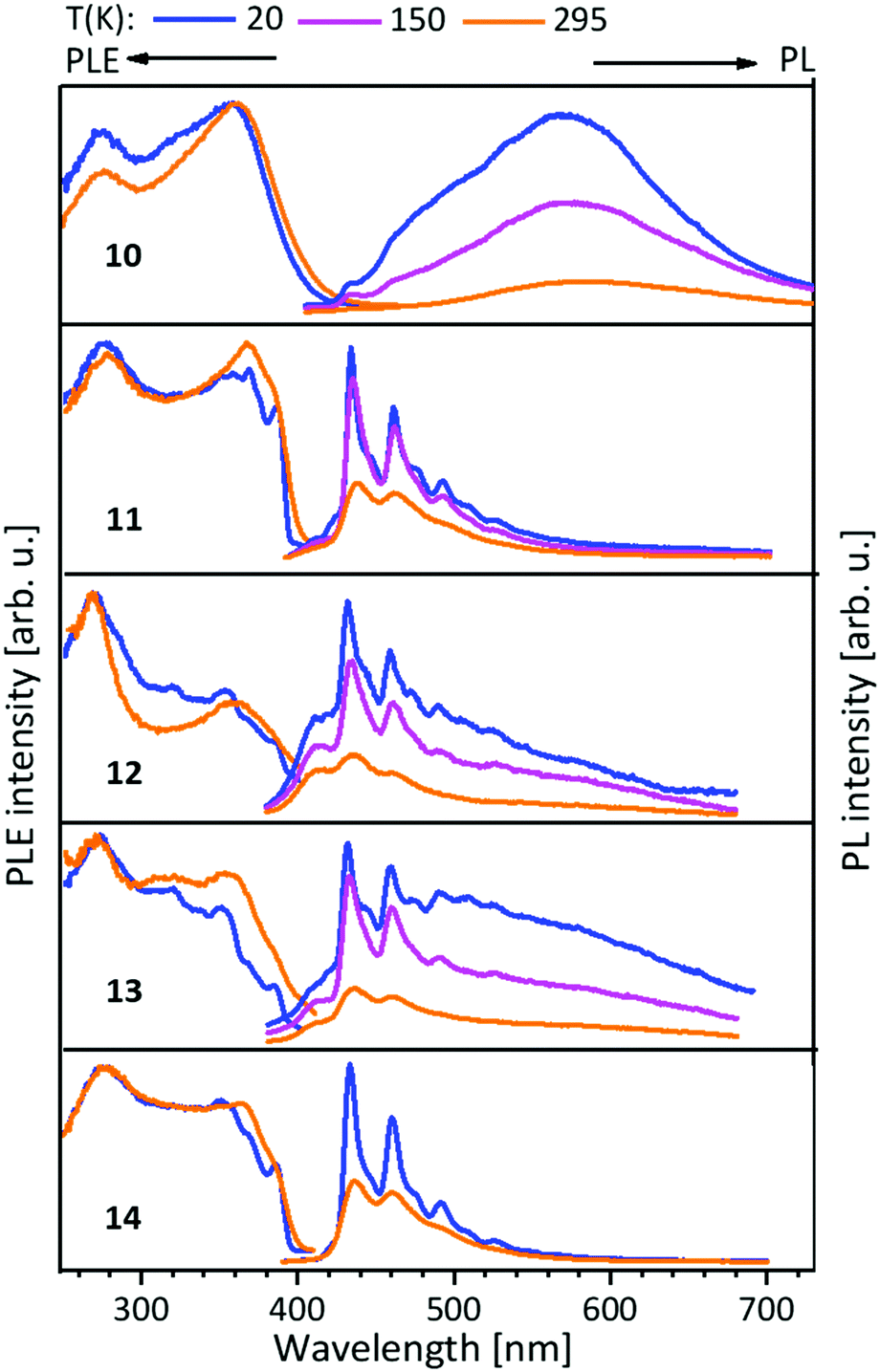 | ||
| Fig. 7 Solid state excitation and emission spectra of NHCimpy (L3) metal complexes 10–14. PL was excited at λexc = 350 nm and PLE spectra recorded at λem = 580 nm for 10 and λem = 430 nm for 11–14, λem = 430/460 nm (13).43 | ||
The PL properties of compounds 1–14 can be finely tuned by the loaded metal, in particular the structural homologue AuCu and AuAg complexes demonstrate different emission spectra. Moreover, the PL efficiency depends on the ligand framework.
In this case L3 coordinated compounds show a much weaker emission,43 especially at lower temperatures, than L1 ligated compounds.41 The structural flexibility of the imino-pyridine function may lead to efficient non-radiative relaxation and PL quenching.43
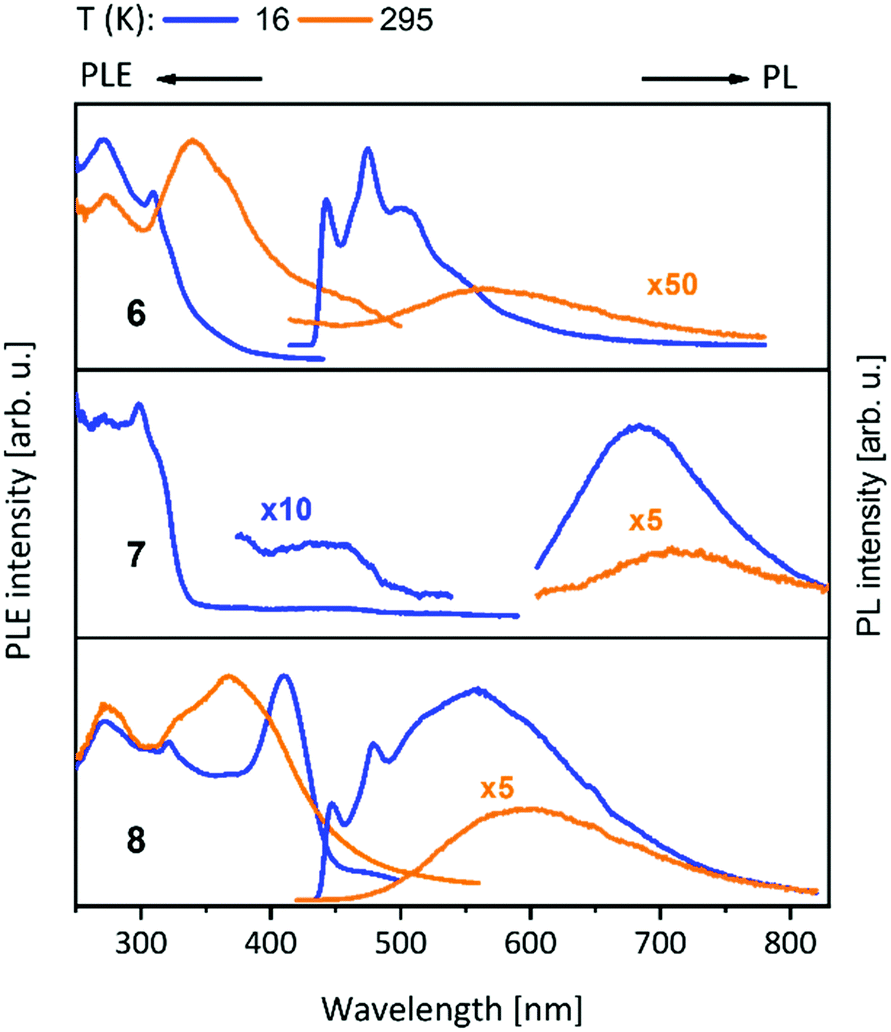
Compounds 15–23 (Table 1) having closely related ligands (L4–L6) (Schemes 6 and 7) show no vibronic structure in their spectra.20,22,45–47 The NHCpy (L4) compound 15 bearing gold(I) and silver(I) shows a sharp band at 416 nm. Exchange of silver(I) by gold(I) in 16 leads to a small shift to 423 nm at 77 K. At room temperature, a broad band at 475 nm can be seen for compound 16.47 NHCpy2 (L5) compounds 17–19 show similar spectra with the band maximum shifted following the Au(I)–Cu(I) separation (Br < I < Cl) with the shortest distances having the highest energy band (509 nm in 18, 514 nm in 19 and 517 nm in 17 at 77 K). The polymeric bromine compound 20 exhibits red shifted emission at 533 nm.46 The almost identical compounds 21 (L5) and 22 (bzNHCpy2 (L6)) feature identical narrow emission bands at 461 nm (21) and 462 nm (22).20,22 The trinuclear complex 23 exhibits strong bright blue luminescence at 455 nm, with enhanced intensity of the emission band when compared to the mononuclear gold(I) precursor [(NHCpy2)2Au][BF4].45
The PL data of compounds 15–23 indicate a clear trend in the energy of the emission peaks by changing the metal loading. Gold(I), silver(I) containing compounds experience a hypsochromic shift, while copper(I) containing compounds are red shifted. Also, the importance of metallophilic interaction on the PL properties can be shown nicely by compounds 17–19 (Table 1).
P–N coordination
Coordination modes
Another approach for ligand systems, capable of multidentate coordination with soft as well as hard donor functionalities, can be achieved by combining phosphorus and nitrogen donor sites. Despite the number of other P–N ligand systems,13,50–53 we focused on papers in which correlation with luminescence properties are considered. Gimeno and co-workers have used bifunctional phosphine ligands (L7, L9 and L10) with pyridine in the backbone in 2010 to access heteronuculear complexes 24, 27 and 28. On reacting the mononuclear gold(I) complexes with [Cu(MeCN)4](X) (X = BF4 or PF6) in 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 molar ratio led to the isolation of trinuclear complexes depicted in Scheme 8.13 The molecular structure of complex 24 in the solid state is established by X-ray analysis revealing a Au2Cu core supported by two ligand scaffolds with an aurophilic (Au1–Au2) interactions of 3.0658(5) Å and Au–Cu contacts in the range of 2.98–3.05 Å.13 Catalano and co-workers synthesised compound 25 with gold(I) coordinated by C6Cl2F3, with parameters similar to those of the closely related chloride compound 24.54
:1 molar ratio led to the isolation of trinuclear complexes depicted in Scheme 8.13 The molecular structure of complex 24 in the solid state is established by X-ray analysis revealing a Au2Cu core supported by two ligand scaffolds with an aurophilic (Au1–Au2) interactions of 3.0658(5) Å and Au–Cu contacts in the range of 2.98–3.05 Å.13 Catalano and co-workers synthesised compound 25 with gold(I) coordinated by C6Cl2F3, with parameters similar to those of the closely related chloride compound 24.54
Au(I) ions are coordinated to a phosphine group and the charge on the ion is balanced by the coordinating chloride. As expected, copper(I) is bonded to the nitrogen atoms of the pyridine moieties. Au(I) and Cu(I) adapt almost linear geometry.13 Complex 27 is also proposed to have a Au2Cu core supported by two ligand scaffolds due to its orange colour which can be ascribed to metallophilic interactions. It is likely that the copper(I) ion is coordinated to four nitrogen atoms thereby resulting in tetrahedral geometry which is different from 24.13 Complex 27 is also comprised of two ligand units and the metal ions defining similar coordination as 24.13 However, in compound 27, the long alkyl linker between pyridine and phosphine groups inhibits metal–metal interactions to exist. The proposed coordination modes in complex 27 and 28 are supported by 1H and 31P{1H} NMR experiments.13 Hobbollahi et al., reported a tri-nuclear complex 26 with L8 and Au2Cu core in 2017 with similar bonding modes to that of 24 which exhibits cold-white emission (Scheme 9).51
Catalano and co-workers have also explored the luminescence properties of heteronuclear complexes synthesised from pyridine-substituted phosphine ligands.55 Complex 29 was synthesised by reacting the Ppy ligand L11 with Au(tht)Cl in 2:1 stoichiometric ratio followed by salt metathesis of the counter-ion using NaBF4.55 Reacting the gold(I) complex 29 with [Cu(MeCN)4](BF4) in acetonitrile yielded complex 30, which on further exposure to air resulted in the formation of complex 31.55 Complex 31 can also be crystallised by slow vapor diffusion of diethylether into a dichloromethane solution of complex 30. Crystals of complex 30 were grown by slow diffusion of diethylether in acetonitrile solution. As expected, the gold(I) ion is coordinated to two phosphorous atoms and adapts a linear geometry in complex 30 and the copper(I) center features a distorted tetrahedral geometry by bonding to nitrogen atoms of two ligand units and two acetonitrile solvent molecules.55 N–Cu–N bond angles vary between 97.68(6) to 138.68(6)°. Complex 31, which is formed due to desolvation of complex 30, varies from 30 in terms of geometry of the metal ions. Complex 31 features short Au–Cu distance of 2.886(2) Å when compared to complex 30 with Au–Cu distance of 3.3736(3) Å. Both gold(I) and copper(I) ions feature almost linear geometry wherein they are coordinated to two phosphorous and nitrogen atoms respectively. Complex 30 exhibits green luminescence which on desolvation demonstrates yellowish luminescence.55
The PN4 ligand system (L12; Scheme 10), in which two bipyridine moieties are attached to a central phosphorus atom fulfils the prerequisites mentioned above,48 just as the monoanionic PNNP ligand (L13; Scheme 11), introduced in 2002 by Tsukada et al.,56 composed of a negatively charged amidinate as nitrogen center with two attached phosphine moieties.57,58
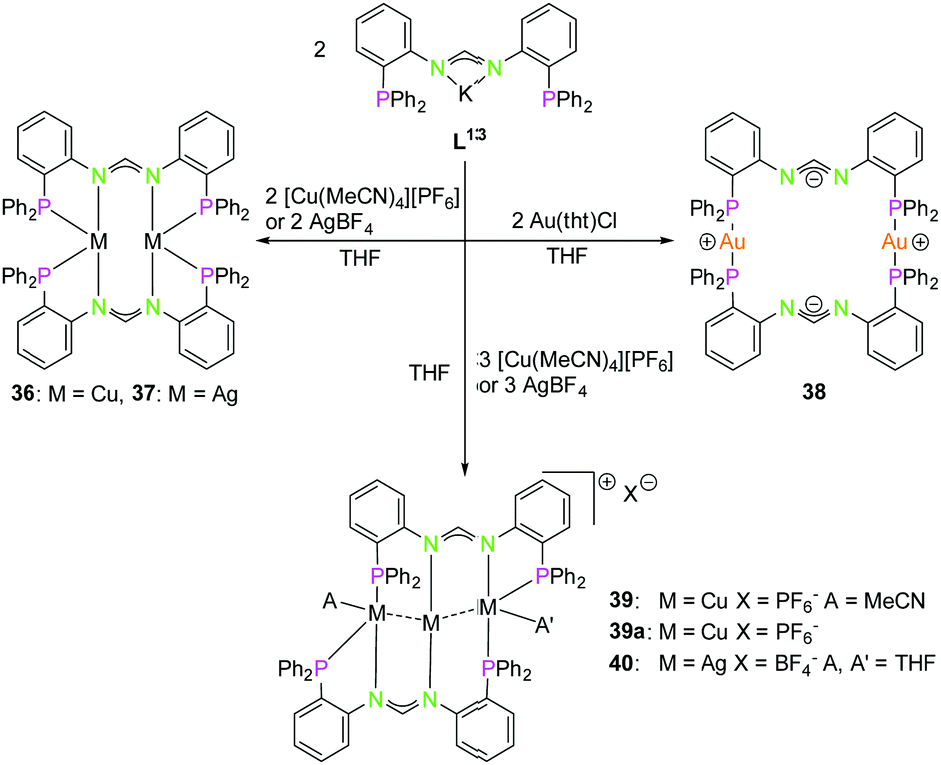 | ||
| Scheme 11 Synthesis of di- (36–38) and trinuclear (39, 39a, 40) homometallic PNNP (K–L13) compounds.58 | ||
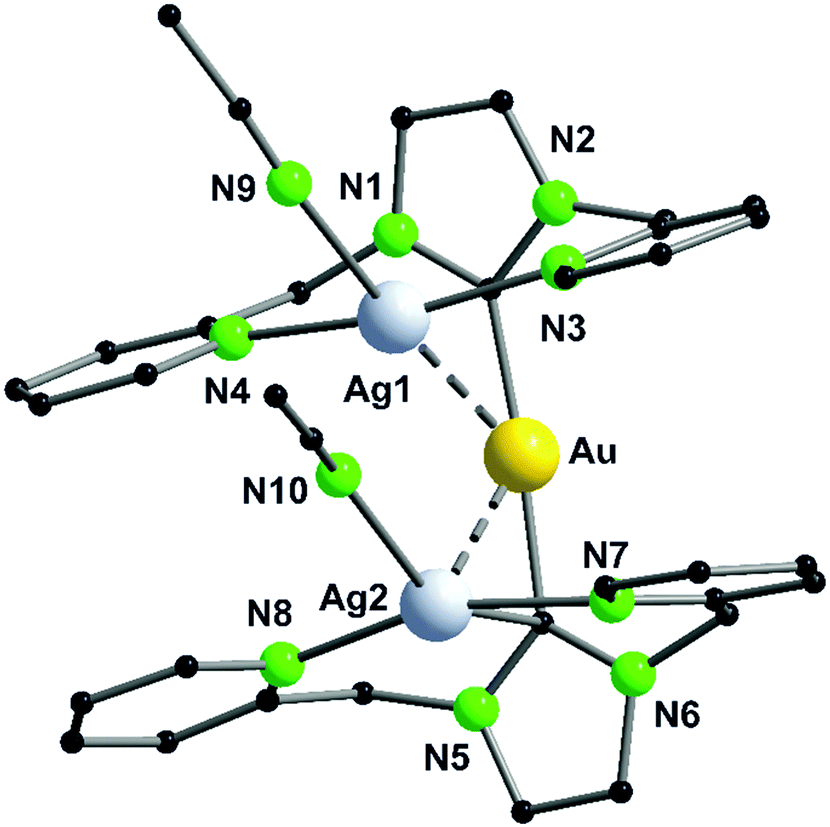
As expected, the reaction of L12 with a gold(I) precursor in a 2:1 ratio leads to the formation of a gold(I) complex linearly coordinated by two phosphorus atoms (32, Scheme 10). The stoichiometric addition of copper(I) (33) and silver(I) (34) precursors leads to trinuclear heterobimetallic complexes, filling the empty coordination compartment of the bipyridine moieties (Scheme 10).48 Both complexes resemble each other, forming an almost linear arrangement of the three metal ions (ca. 179°), with tetrahedrally distorted coordinated silver(I) or copper(I) atoms. These two compounds show metallophilic interactions, having Au–M distances of around 3 Å for silver(I) as well as copper(I), implying that the range of the metal–metal distances are mainly a consequence of the rigid ligand geometry.59–61
A trinuclear homometallic silver(I) complex 35 was synthesized, displaying the identical structural arrangement as the two heterometallic compounds 33 and 34. Here, silver(I) is coordinated by a significant more distorted tetrahedral geometry highlighted through the varying length of Ag–N bonds, which is greater than the corresponding gold(I)–silver(I) complex (Fig. 8).48,62
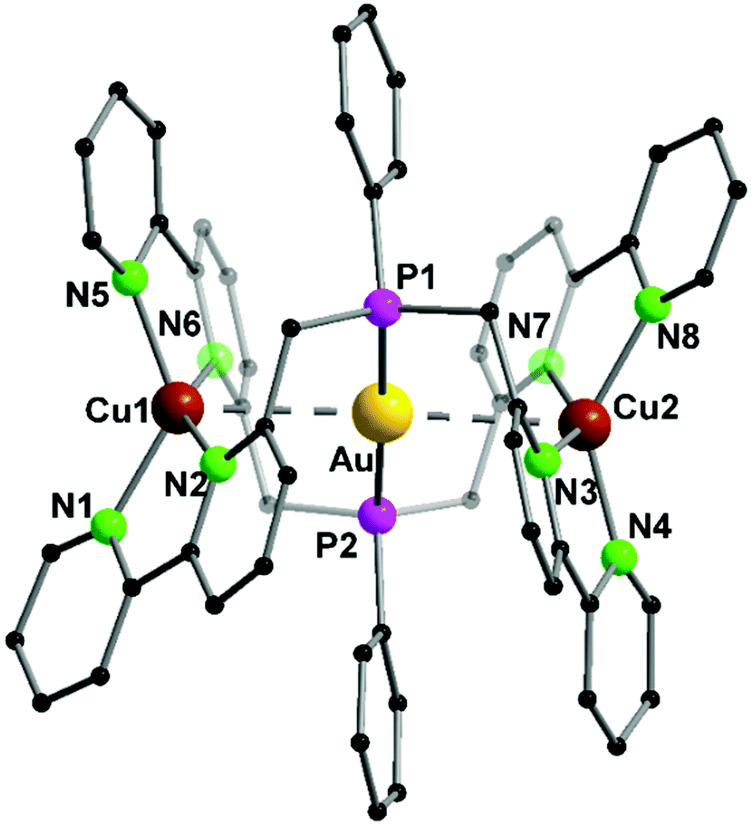 | ||
| Fig. 8 Molecular structure of 33 in the solid state. Hydrogen atoms and the counter ions are omitted for clarity.48 | ||
The reaction of coinage metal precursors with the potassium salt of the amidinate based PNNP ligand L13 (Scheme 11) leads to neutral dinuclear compounds, forming isostructural complexes with copper(I) (36) and silver(I) (37).58 In contrast to L1–L12, ligand L13 is an anionic ligand, which leads to metal complexes with an overall lower charge. In general, these are better soluble in organic solvents than their highly charged counterparts 32–34. The metal atoms in 36 and 37 are each coordinated by two nitrogen atoms and two phosphorous atoms in a distorted tetrahedral environment. Although bis(amidinate) copper(I) complexes similar to 36 are known to form short Cu–Cu contacts, no cuprophilic interaction is observed (3.63 Å) in 36,63,64 most likely since the phosphine moiety allows the more favoured tetrahedral coordination. In contrast an intermetallic distance of 3.44 Å indicates weak argentophilic interactions for the dinuclear silver(I) complex 37 in good agreement with literature values.9
Despite literature known dinuclear gold(I) bis(amidinate) compounds with gold(I) coordinated by two nitrogen atoms,65,66 the structure of 38 is differing significantly from these and from structures with the lower homologues.
A slightly bent bis(phosphine) gold(I) coordination mode is obtained, showing the preference of gold(I) for soft donors and low coordination numbers. A remarkable charge separation was observed for compound 38, with the negative charges delocalised over the NCN and the positive charges situated on the gold(I) atoms.58,67 With a ligand to reactant ratio of 2:3 it is possible to synthesise trinuclear homometallic complexes (39, 40), revealing a tilted Cu3 chain (117.84(1)°) (Scheme 11). All three copper(I) atoms are in different coordination modes, not taking the cuprophilic interactions into account. Cu1 is almost trigonal planar coordinated, Cu2 is almost linearly coordinated and Cu3 is additionally coordinated by one molecule of acetonitrile, resulting in a distorted tetragonal coordination.
Due to the different coordination geometries of the metal ions intermetallic distances are varying, leading to a short 2.5984(4) Å (Cu1–Cu2) and a longer 2.7792(4) Å (Cu2–Cu3) interaction.8,58 Crystallisation in absence of MeCN leads to a similar scaffold with a bent Cu3 chain (39a) (122.07(3)°) and both outer copper(I) atoms are distorted trigonal planar coordinated. This results in a roughly similar Cu–Cu distance (ca. 2.56 Å) between the outer and the central copper(I) atoms. Finally, the homologue silver(I) complex was synthesised (40), showing a weakly bound THF at both outer silver(I) atoms, giving a more symmetrical arrangement than for the copper(I) (MeCN) compound 39, with the Ag3 chain slightly more bent (133.861(10)°) and intermetallic distances around 2.89 Å.9,58
The dinuclear gold(I) compound 38, mentioned above (Scheme 11), still has open coordination sites and addition of coinage metal precursors resulted in three tetranuclear coinage metal chain complexes (41–43) (Scheme 12).57 The inner lying atoms of the heterometallic complexes (41, 42) are each coordinated by two nitrogen atoms and one THF molecule forming a trigonal planar coordination or, if the intermetallic interactions are taken into account, a distorted trigonal bipyramidal coordination. The gold(I) atoms in all three compounds are almost linear coordinated by two phosphorous atoms and almost planar T-shaped considering the Au–M contact, respectively. In contrast to the heterometallic compounds, the inner gold(I) atoms of the tetranuclear homometallic compound 43 are not coordinated by solvent molecules consequently having an almost linear coordination. The average intermetallic angle M–M–M is almost identical for the copper(I) and silver(I) compound (121.92° Cu; 119.42° Ag) but significantly widened for gold(I) (131.63°).57
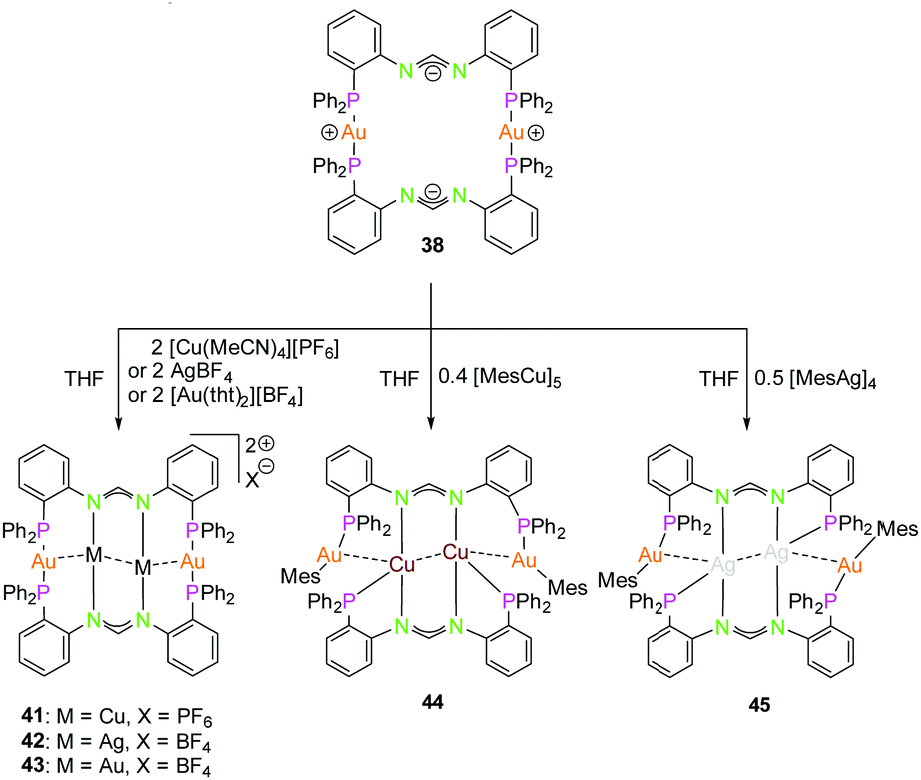 | ||
| Scheme 12 Synthesis of tetranuclear hetero- (41, 42, 44, 45) and homometallic (43) PNNP (L13) compounds.57,68 | ||
It should be mentioned, that even through gold(I) prefers soft donor sites, a coordination by hard nitrogen donors is possible with enough stabilisation.66,69 A trend can be observed for the intermetallic distances. The outer Au–M contacts are in a range of 2.8 Å to 2.9 Å with the order Ag(I) > Au(I) > Cu(I), also similar for the inner metallophilic interaction (2.7334(2) Å Ag; 2.6998(8) Å Au; 2.5995(3) Å Cu). This shows, that even though gold(I) has the greater van der Waals radii than silver(I), it forms stronger interactions with smaller intermetallic distances.57,70–73 The metalloligand 38, when reacted with mesityl copper(I) and mesityl silver(I) in THF, resulted in the tetranuclear heterometallic complexes 44 and 45 in 36% and 33% yield respectively (Scheme 12).68 Both complexes were crystallised from THF and n-pentane. Molecular structure of 44 in the solid state revealed that each inner lying copper(I) atom is coordinated to two nitrogen atoms of the amidinate group and to a phosphine unit in an almost T-shaped geometry.68 The metals arrange themselves in a Au–Cu–Cu–Au zigzag chain with an angle of 111.62(2)° and intermetallic distances of 2.7939(9) Å (Cu–Cu) and 2.9960(4) Å (Au–Cu) which are in the range of metallophilic interactions. Following the HSAB principle, gold coordinates to phosphorous and a mesityl unit. Additionally, gold deviates from its usual linear geometry with an angle of 167.67(10)° (P–Au–mesityl).68 Complex 45 also demonstrates a rather similar tetranuclear zigzag chain Au–Ag–Ag–Au wherein the intermetallic bond distances are 2.9756(3) Å (Au–Ag) and 2.9009(5) Å (Ag–Ag) and the bond angle is 102.197(13)°.68
In contrast to Au–Cu complex 44, the mesityl groups which are exchanged from silver(I) to gold(I) atoms are located on the opposing ligand (Scheme 12). Each gold(I) atom in complex 45 is coordinated to a phosphorous and a mesityl unit, thereby adapting an almost linear geometry (173.80(10)°). Similar to complex 44, the inner lying silver(I) atoms are coordinated by two nitrogen atoms and a phosphorous moiety and when metallophilic interactions are taken into consideration, silver(I) atoms are in an overall trigonal bipyramidal coordination environment. However, due to low solubility of the complexes, NMR investigations are not reported (Fig. 9).68
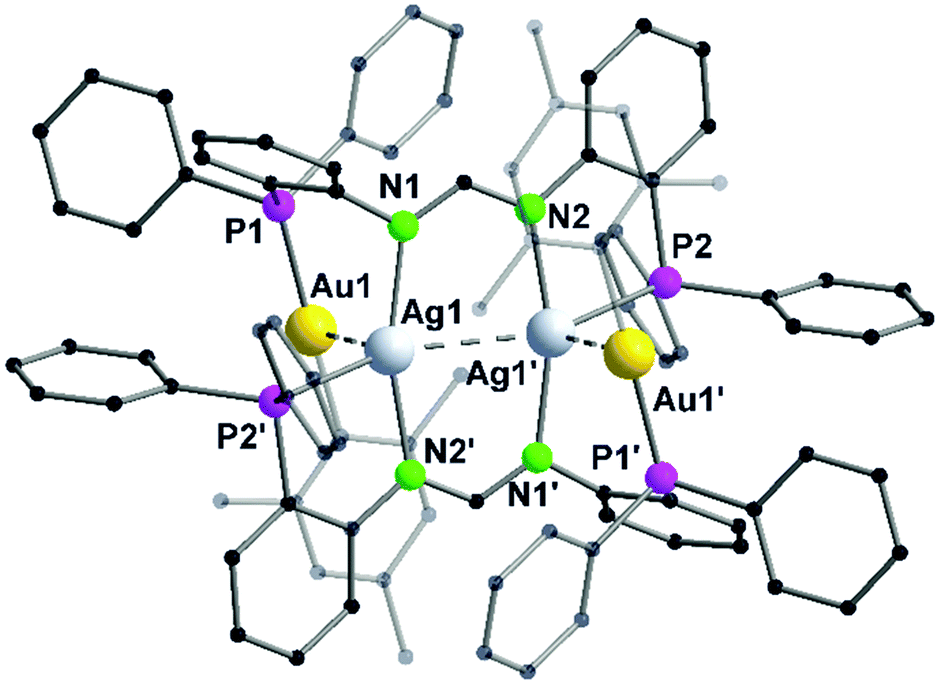 | ||
| Fig. 9 Molecular structure of 45 in the solid state. Hydrogen atoms and the counter ions are omitted for clarity.68 | ||
Scheme 13 displays several P–N ligands L14–L18 containing a diphenyl phosphine unit as a soft donor and a phenanthrene (L14), aniline (L15), napthtyridine (L16), imidazole (L17), or a triazole (L18) function as hard nitrogen donor. Their complexes 46–51 can be synthesised straight forward by addition of the corresponding coinage metal precursors in a stoichiometric manner.34,50,54,74,75 Complex 46 consists of two gold(I) ions, coordinated by the diphenyl phosphine and a C6Cl2F3 in an almost linear fashion, and a copper(I) core coordinated by the two nitrogen atoms of phenantrene and an acetonitrile molecule in a distorted trigonal conformation. The rigid ligand geometry prevents the close association of the metal centers, thus no metal interactions in the solid state can be noted.54 The dinuclear compounds 47 and 48 are closely related. Herein, the gold(I) is coordinated by the two phosphorus atoms in a linear manner, and the second metal ion by two nitrogen donors. The Au–Ag distance is ca. 3.0 Å.74 Compound 49 shows a trigonal coordination of both metal ions gold(I) and copper(I) forming almost parallel plains (P3Au, N3Cu). Copper is coordinated by the second nitrogen of the napthtyridine, thus resulting in a long Au–Cu distance of 4.47 Å.34 Similar to compound 49, complex 50 has trigonal coordinated metal ions. The highly symmetrical compound 50 has intermetallic interactions with a distance of 2.86 Å (Au–Ag). Experiments to synthesise the linear coordinated species with ligand L17 were unsuccessful and only works for the homometallic gold(I) and silver(I) compounds.50 At last, compound 51 having a similar solid state structure as 24 and 26, but with a slightly elongated Au2Cu core (Au–Au 3.48 Å, Au–Cu 3.06–3.20 Å) and copper(I) is additionally coordinated by two thf molecules.13,75 Interestingly the ligand L18 can not only be used as a P–N orthogonal ligand but also as P–C ligand by alkylating the nitrogen donor and deprotonation to the mesoionic carbene species or by deprotonation to the triazolide. The resulting soft–soft coordination sites are capable of building dinuclear homometallic gold(I) compounds.75
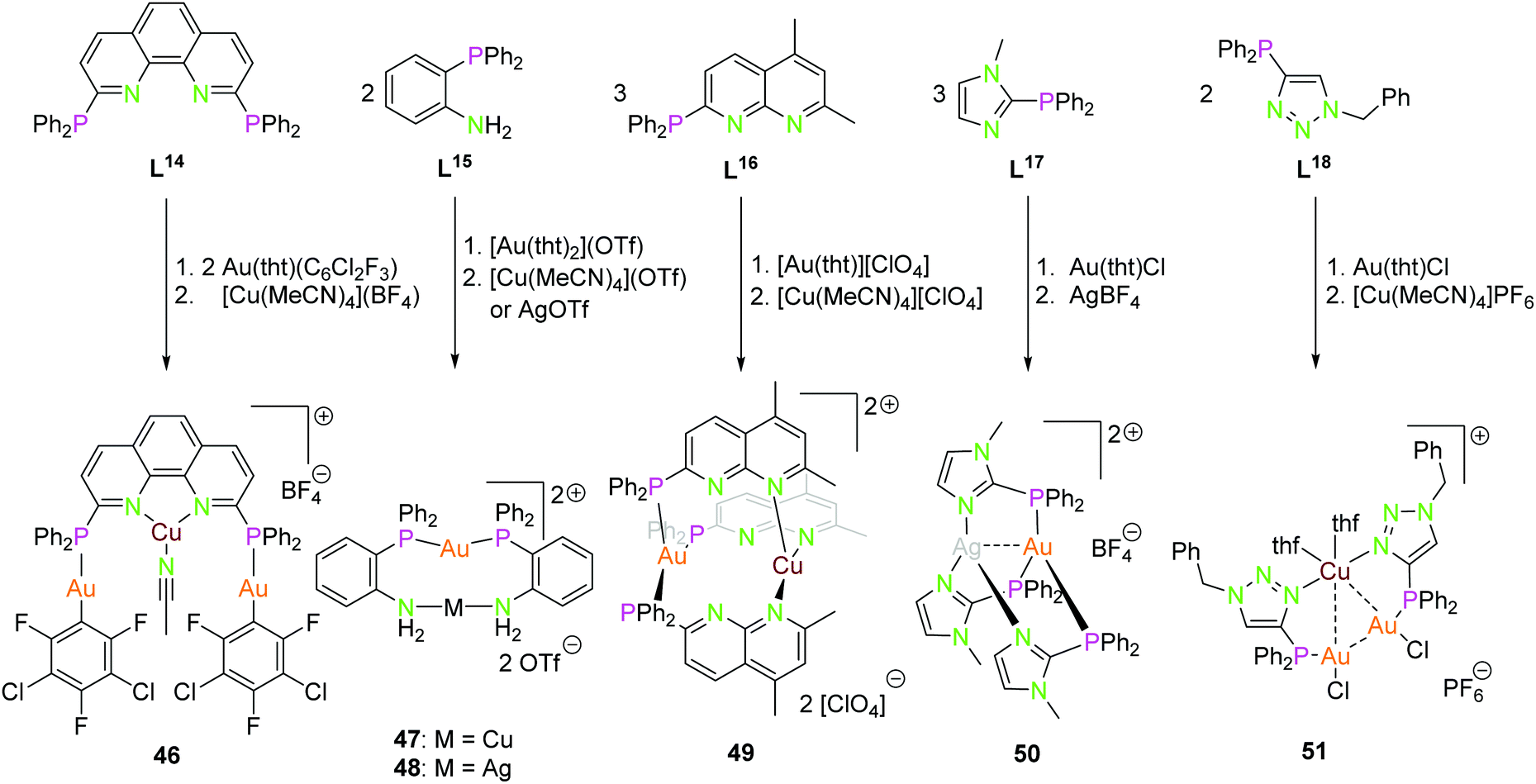 | ||
| Scheme 13 Synthesis of heterometallic di- (47–50) and trinuclear (46, 51) compounds with ligands L14–L18.34,50,54,74,75 | ||
The structures depicted in Scheme 13 show compounds with trigonal planar coordinated (46, 49, 50) as well as linear coordinated (46–48) coinage metals. In particular, compounds 47, 48 and 50 show that linear or trigonal coordination can be modulated by the ligand system, respectively.
Depicted in Scheme 14 is the synthesis of the tetranuclear bimetallic gold(I)–copper(I) compound 53 using the (PPh2)2Py ligand L19. Simple addition of 2 equiv. of Cu(SMe2)Br, followed by silver(I)triflate leads to the dinuclear copper() compound 52, which can be converted to 53 by transmetalation with Au(SMe2)Cl. Compound 53 has linear coordinated gold(I) ions bridging the two ligand moieties, copper(I) rises above and below the paper plane and is linear coordinated by the pyridine nitrogen and one chloride. The gold(I)–copper(I) intermetallic distance is between 3.034 and 3.065 Å and the Au2Cu2 core forms a tetranuclear planar shaped metal alternating tetragon.18
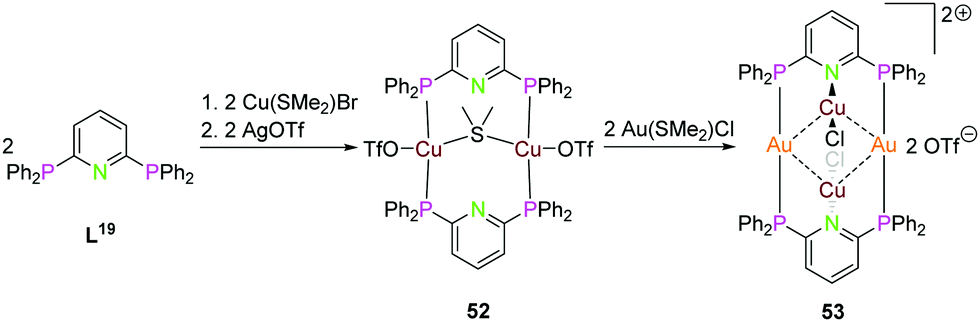 | ||
| Scheme 14 Synthesis of homo- (52) and heterometallic (53) compounds with ligand L20.18 | ||
Like above-mentioned for C–N coordination, the combination of soft phosphorous and hard nitrogen donor sites also allows a selective coordination of coinage metals. The advantage of these selectivity is reflected by the changes in the PL properties with different metal loadings and coordination spheres.
Photoluminescence properties
Metalloligands L7–L10 do not exhibit luminescence at ambient temperature. However, on introducing copper(I) as a hetero metal, the complexes exhibit different emissive behaviour.Complexes 24–27 (Table 2) in the solid-state exhibit broad emission band centered at 516 nm, 542 nm, 450 nm (broad shoulder at approx. 550 nm) and 558 nm at room temperature respectively.47,51 The lifetimes for these complexes 24 and 27 at room temperature are measured to be 29 μs and 13 μs respectively indicating phosphorescent nature.13 The emission of complex 26 at 297 K is also phosphorescent in nature, resulting from minimum of two non-equilibrated triplet states with lifetimes of τ450nm = 2.2(77)/6.3(23) μs and τ550nm = 12 μs (pre-exponential factors are mentioned in brackets). In contrast to complexes 24–27, the emission band of 28 appears at lower energy, which is attributed to the different coordination geometry of copper(I) (tetrahedral).13 Heterometallic complex 30 shows blueish emission in acetonitrile solution (λmax = 367, 389 nm (shoulder)) and intense emerald colour (λmax = 412 nm) in the solid state.55 Complex 31 which formed due to loss of acetonitrile solvent molecules from 30 features thermochromism behaviour (i.e., exhibits different emission colours at different temperatures) in the solid-state. It emits intense, yellow-coloured emission at 298 K, with the emission band centred at λmax = 551 nm and when heated to 343 K, λmax is shifted to 536 nm (aquamarine), due to endothermic phase transition and remains unchanged above 343 K. On cooling the sample to 77 K the emission is green coloured with λmax = 543 nm, which is attributed to rigidochromism (mechanical sensitivity). The thermochromism behaviour is reversible and is maintained for more than five heating/cooling cycles (Table 2).55 The PN4 compounds 32–35 (Scheme 10) show roughly similar broad emission with a slightly visible vibronical structure for the mononuclear gold(I) complex 32 with the highest intensity at 520 nm (Fig. 10). The heterometallic trinuclear gold(I)–silver(I) complex 34 has a bathochromic shift to 580 nm and the gold(I)–copper(I) compound 33 is shifted further to 740 nm in the NIR region due to the HOMO contribution from the d-orbitals of copper(I). Also, the PL onset at about 720 nm leads to the deep-red colour, while the other trinuclear compounds 34 and 35 have a PLE onset at 400–450 nm. The replacement of gold(I) with silver(I) in the homometallic trinuclear silver(I) complex 35 shows no impact on the PL properties (Fig. 10).48 PL spectra of dinuclear compounds 36–38 (Scheme 11) look quite identical, with similar PLE spectra onset at ca. 450 nm (Fig. 6). The vibronically structured emission is similar for copper(I) (36) and silver(I) (37), with emissions around 520 nm, but since the gold(I) compound 38 has a structural difference, the PL spectra shows a distinct spectrum with a blueshift of the emission (Fig. 11).58 Both trinuclear complexes of copper(I) 39 and 39a (Scheme 11) show similar PLE spectra with onset at 450 nm and PL emission at ca. 500 nm, but the shape of the PL spectra is different (Fig. 6). The Cu(MeCN) complex 39 shows a vibronic pattern, while it is absent without coordinating solvent (39a), leading to the conclusion that the vibronic “modulation” can be attributed to the acetonitrile coordination. Accordingly, the silver(I) compound 40 (Scheme 11) with coordinating THF molecules shows no vibronic pattern but is significantly blueshifted towards 460 nm with a PLE onset at 360 nm (Fig. 11).57,58Fig. 12 shows the PL spectra of the tetranuclear complexes 41–43 (Scheme 12), all having broad (especially the copper(I) compound 41) emission maxima, with a significant bathochromic shift from silver(I) (42) (430 nm) to gold(I) (43) (490 nm) to copper(I) (41) (530 nm). This bathochromic shift of copper(I) and blueshift of silver(I) relatively to gold(I) is in overall accordance with the behaviour of the other described compounds when the coinage metal composition is changed.57
| λ ex (nm) | λ em (nm) | τ obs (μs) | Φ em (%) | |
|---|---|---|---|---|
| 298 K | 298 K | 298 K | 298 K | |
| 24 | 320, 360 | 558 (+sh) | 29 | — |
| 25 | 390 | 542 | 2.82 | 14 |
| 26 | 350 | 450, 550 (sh) | 2.2(7.7)/6.3(23) 12 | 28.0 |
| 27 | 360 | 516 (br) | 13 | — |
| 28 | 460 | 715 | — | — |
| 30 | 350 | 536 | — | — |
| 31 | 350 | 551 | — | — |
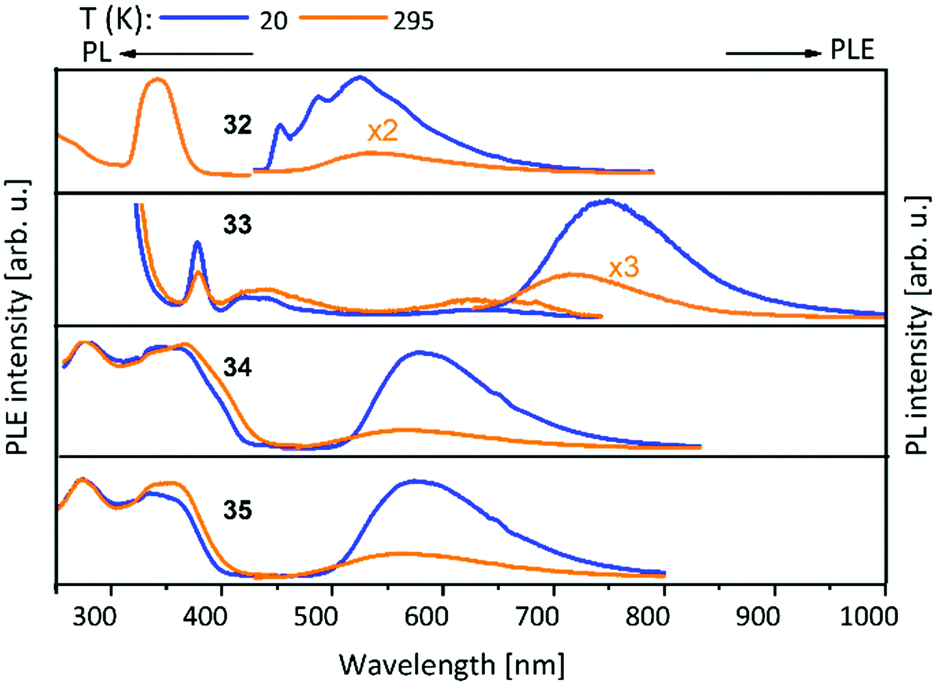 | ||
| Fig. 10 Solid state excitation and emission spectra of PN4 (L12) metal complexes 32–35. PL was excited at λexc = 350 nm and PLE spectra recorded at λem = 560 nm for 32, 33, 35 and λem = 580 nm for 34.48 | ||
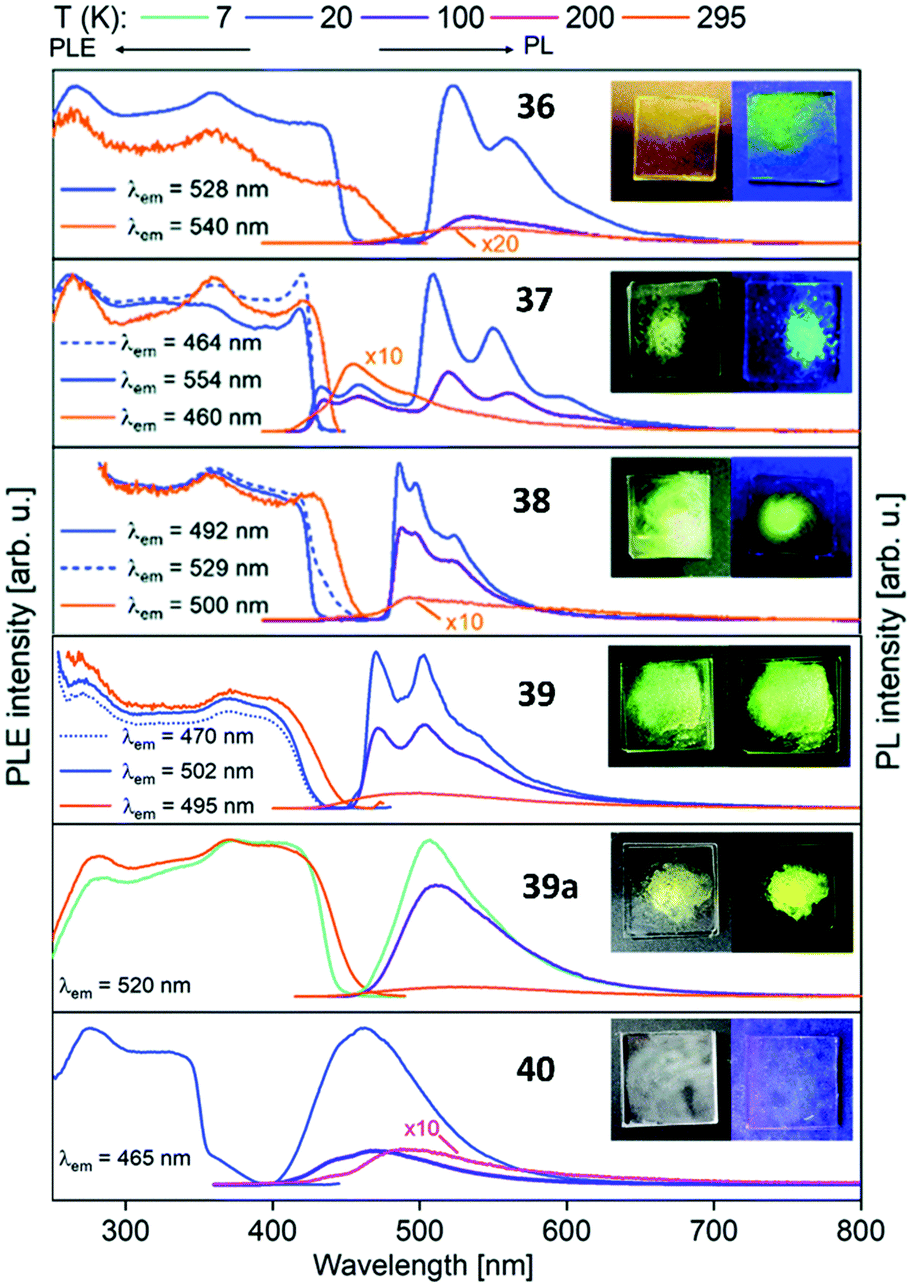 | ||
| Fig. 11 Solid state excitation and emission spectra of di- and trinuclear PNNP (L13) metal complexes 36–40. PL was excited at λexc = 350 nm for 36–39a and λexc = 330 nm for 40.58 | ||
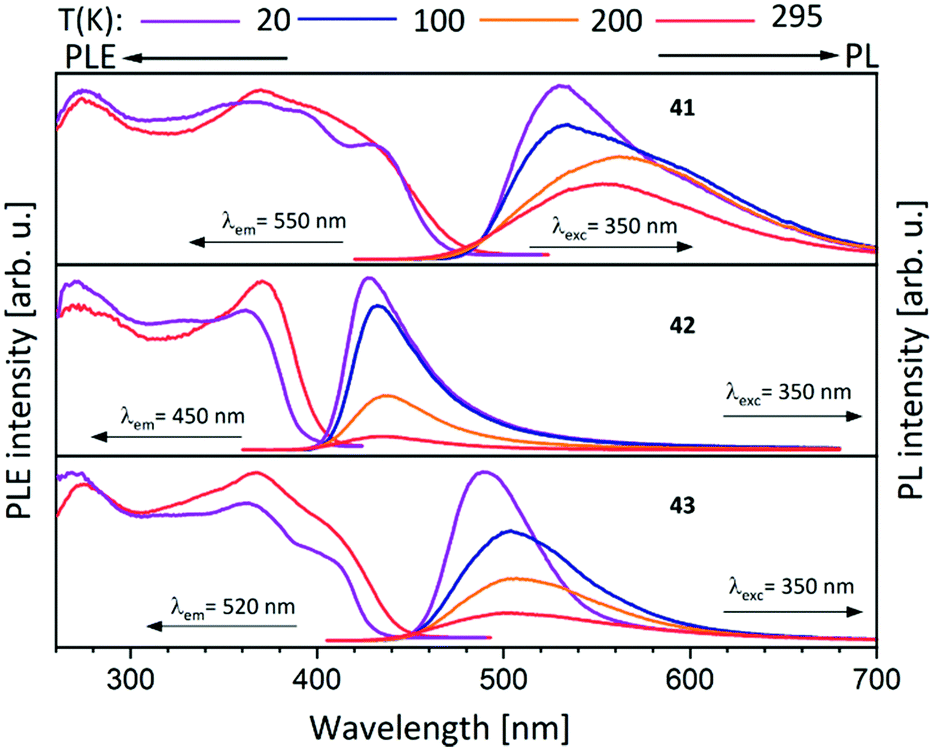 | ||
| Fig. 12 Solid state excitation and emission spectra of tetranuclear PNNP (L13) metal complexes 41–43.57 | ||
Complexes 44 and 45 exhibit broad emission bands both at ambient and low temperatures in the solid-state with a maximum at 600 nm and 482 nm respectively (Table 3).68 Like previously discussed complex 33,48 a bathochromic shift in the PL spectra is observed for the copper(I) complex 44.68 The quantum yields of compounds 44 and 45 were reported to be 35% and 2% respectively and the PL efficiency reaches nearly 100% at temperatures below 100 K (Table 3).68 Complexes 44 and 45 are phosphorescent, and the lifetimes are measured to be 310 μs for 44 and ca 320 μs for 45 at 20 K. However, the lifetimes tend to decrease with an increase in temperature.68 Additionally, we want to mention the observation of a bathochromic shift for compounds 41 and 43 when measured in the gas phase, solution and solid state. TDDFT calculations showed, that the electronic properties of Au–M–M–Au complexes can be strongly modulated by metallophilic Au–M interactions. The remarkable agreement of experimental and calculated energies of ionic compounds in gas phase demonstrate the importance of gas phase PL spectroscopy combined with quantum chemical calculations.57
| λ ex (nm) | λ em (nm) | τ obs (μs) | Φ em (%) | ||
|---|---|---|---|---|---|
| 298 K | 298 K | 298 K | 77 K | 298 K | |
| 44 | 350 | 600 | — | 310 | 35 |
| 45 | 350 | 482 | — | 320 | 5 |
| 46 | 455 | 615 | 2.7/0.7 | — | 8 |
| 50 | 360 | 490 | — | — | — |
| 51 | 350 | 580 | 8 | 14 | 9 |
| 53 | 334 | 490 | 7.7 | 12 |
Phenantrene compound 46 shows a broad asymmetric band at 615 nm, with biexponential decay times τ = 2.7/0.7 μs and a quantum yield φ(295 K) of 8% in the solid state (Table 3). In THF solution a structured emission at 436 nm can be observed (Table 4), possibly due to the coordination of solvent, like for compound 39.54,58 Compounds 47 and 48 both have featureless bands at 431 nm (Au–Cu) and 430 nm (Au–Ag) (Table 4).74 The trigonal gold(I)–copper(I) compound 49 exhibits an emission band at 530 nm and is blueshifted in comparison to the mononuclear gold compound (560 nm) (Table 4).34 The second trigonal compound 50 shows a broad featureless band at 490 nm in the solid state and a blueshifted single emission band at 354 nm.50 Compound 51 displays a broad featureless emission band centered at 580 nm, typical for charge transfer transitions. The emission decay are τ = 14 μs and 8 μs at 20 K and 295 K, respectively and the quantum yield φ(295 K) is 9% (Table 3).75 Compound 53 shows abroad peak at 490 nm with a lifetime of 7.7 μs and a quantum yield φ(295 K) of 11.8% (Table 3).18
| λ ex (nm) | λ em (nm) | |
|---|---|---|
| 298 K | 298 K | |
| 46 | 373 | 436 |
| 47 | 350 | 600 |
| 48 | 350 | 482 |
| 49 | 455 | 615 |
P–O coordination
Coordination modes
Following Pearsons‘ concept of HSAB principle, combination of P- and O-donor sites presents an additional class of bifunctional ligands which fulfils the pre-requisite for the synthesis of heterobimetallic complexes, with oxygen being a hard donor, preferring coordination with copper(I) as well as silver(I). Koshevoy and co-workers have reported a series of coinage metal complexes using a mixed phosphine–phosphine oxide (P–PO) hybride ligand.76 However, to the best of our knowledge, there has been no other coinage metal complexes reported with P–O bifunctional ligands, which are studied with respect to their optical properties. The limited study corresponding to this class of ligands is attributed to the challenging synthesis and low selectivity of unsymmetrical oxide derivates when compared to their parent phosphines (Scheme 15).76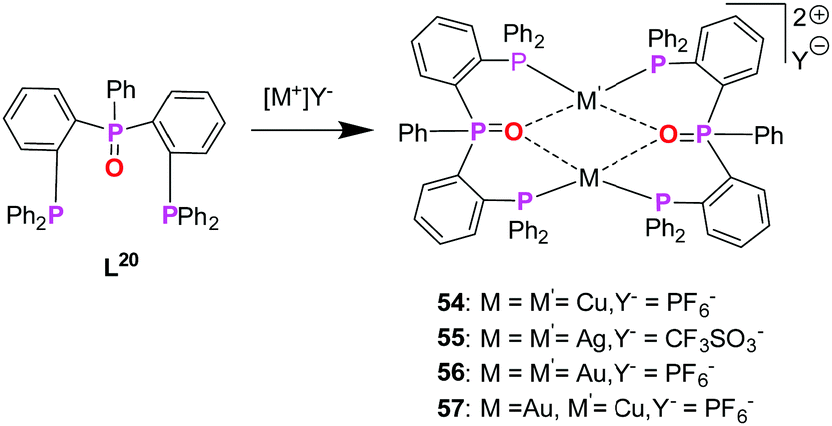 | ||
| Scheme 15 Synthesis of homo- (54–56) and heterometallic (57) complexes based on mixed phosphine–phosphine oxide hybrid ligand (L20).76 | ||
Koshevoy and co-workers were successful to synthesize bis(triphenylphosphine)phenylphosphanoxide (L20) in 80% yield. Ligand L20 was employed for the synthesis of several homometallic complexes and a heterobimetallic complex (Scheme 14).76
The heterobimetallic complex 57 was synthesized by reacting the homometallic complexes 54 and 56 in DCM for 2 h. The complex was isolated as yellow crystalline solid in 84% yield. The molecular structure in the solid state was established using single crystal XRD analysis. L20 being a tridentate ligand, provides two types of ligand spheres in the complex 57 (“P2O2” and “P2”). The ligand sphere P2O2 includes a phosphine group and phosphine oxide of two ligand moieties, which accommodates Cu(I) ion and the P2 ligand sphere is comprised of the other phosphine functionality of the ligand, which saturates the coordination vacancy of Au(I). Copper(I) is coordinated by the lone pairs of phosphorous and oxygen atoms, adopting a distorted tetrahedral coordination geometry. Whereas gold(I), coordinated by two phosphorous atoms, slightly deviates from ideal linear geometry, which is attributed to weak Au–O interactions. Silver(I), with its behaviour in between, coordinates only to one oxygen atom and is additionally weakly η2 coordinated to a C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C double bond of a phenyl ring, leading to a strongly distorted tetrahedral coordination.76 The purity of the complexes was established using NMR spectroscopic measurements and elemental analysis. NMR investigation of compound 57 revealed the presence of a dynamic equilibrium in solution between heterobimetallic complex 57 and its homometallic congeners. However, on recrystallisation of the mixture, crystals of complex 57 can be recovered which was characterized by XRD analysis. Additionally, complex 57 shows a unique emission spectrum which is different from its homometallic congeners which supports the analytical purity of 57 (Fig. 13).76
C double bond of a phenyl ring, leading to a strongly distorted tetrahedral coordination.76 The purity of the complexes was established using NMR spectroscopic measurements and elemental analysis. NMR investigation of compound 57 revealed the presence of a dynamic equilibrium in solution between heterobimetallic complex 57 and its homometallic congeners. However, on recrystallisation of the mixture, crystals of complex 57 can be recovered which was characterized by XRD analysis. Additionally, complex 57 shows a unique emission spectrum which is different from its homometallic congeners which supports the analytical purity of 57 (Fig. 13).76
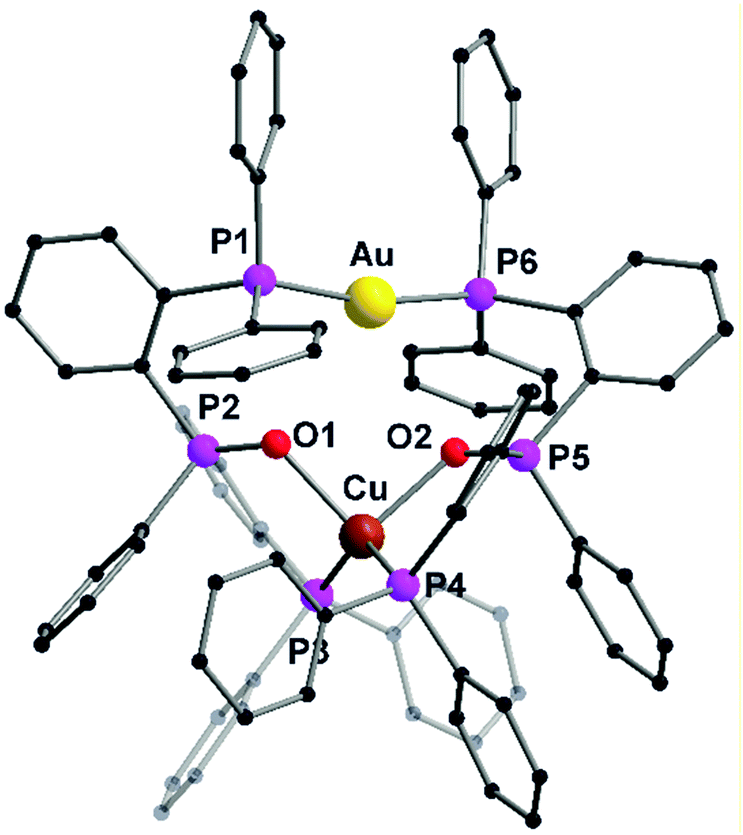 | ||
| Fig. 13 Molecular structure of 57 in the solid state. Hydrogen atoms and the counter ions are omitted for clarity.76 | ||
Photoluminescence properties
The complexes 54–57 (Scheme 14) do not exhibit significant luminescence in solution, hence, only the solid state photophysical properties are summarised in Table 5. Well-reported bathochromic shift for copper(I) complex 54 and hypsochromic shift of silver(I) complex 55 relative to gold(I) complex 56 is observed. Gold(I)–copper(I) complex 57 displays a broad emission band centered at 538 nm. The emission band of 57 is blue shifted with respect to the emission spectra of 54 and 56 which are centred at 604 and 548 nm, respectively. Gold(I) containing complexes 56 and 57 exhibit comparatively larger quantum efficiencies due to increased radiative rate constants. The effect is mainly due to larger spin orbit coupling present in gold(I) complexes and a higher inter system crossing (ISC) rate when compared to their first and second row transition metal complexes.76 The lifetime for complexes 54–57 is in the microseconds range at 298 K which drastically increases on lowering the temperature to 77 K. Heterobimetallic complex 57 doesn’t exhibit any significant difference in terms of quantum yield and lifetime when compared with 54 and 56 at 298 K. However, the lifetime for complex 57 at 77 K is 777.5 μs which is considerably greater than the homometallic analogues 54 (174.1 μs) and 56 (82.3 μs).76| λ ex (nm) | λ em (nm) | τ obs (μs) | Φ em (%) | ||
|---|---|---|---|---|---|
| 298 K | 298 K | 298 K | 77 K | 298 K | |
| 54 | 364 | 604 | 0.35 | 174.1 | 0.5 |
| 55 | 332 | 500 | 10.9 | 2354.3 | 5.9 |
| 56 | 330 | 548 | 6.7 | 82.3 | 28.9 |
| 57 | 360 | 538 | 5.5 | 777.5 | 25.8 |
Conclusions
The selective coordination of transition metals can be achieved by using hard and soft donor sites following the HSAB principle of Pearson. For the selective coordination of several different metals to the same ligand system, specially designed ligand systems are needed. The combination of C–N, P–N or P–O coordination sites in the same ligand, resulting in orthogonal ligands, is one way to achieve this. The specific preferences of the coinage metals allow the synthesis of multinuclear heterometallic complexes, where the metal can be selectively coordinated to different donor sites.Copper(I) prefers hard donor sites, with high coordination numbers, and can be found in tetrahedral, trigonal planar but also in linear coordination modes. The same applies to silver(I) but a propensity to lower coordination numbers and the formation of intermetallic interactions can be noted. Finally, gold(I) as soft metal coordinates to soft donor functions with low coordination numbers. It can be said that all featured ligand systems C–N, P–N and P–O show the expected selectivity and therefore are good examples for orthogonal ligand designs. Besides the fixation of orthogonal donor sites, the ligand architecture is also essential for the nuclearity of the corresponding complexes. We have shown examples ranging from bi- to tetranuclear complexes.
Another outstanding property of the coinage metals is their propensity to form strong intermetallic interactions, known as metallophilicity. This increases the interest in the formation of heterometallic multinuclear coinage metal complexes. With the metals in close range to each other, the influence of the variable metal loading on their coordination behaviour as well as the intermetallic exchange can be investigated.
It must be mentioned that the metallophilic interactions also have an impact on the PL properties and can be easily tuned by exchange of the metal loading. Thus, copper(I) containing compounds usually experience a bathochromic shift while silver(I) tends to a hypsochromic shift.
Overall, there are many ligand systems with C–N and P–N donor sites, but not many findings for P–O orthogonal ligands. Additionally, sulphur, in combination with nitrogen or oxygen presents an excellent class of orthogonal ligands. There are only a few examples of such ligands. Konno et al. synthesised multimetallic homo- and heteronuclear metallorings from benzothiazoline.77 However, the optical properties of the metal complexes were not reported. Other sulphur containing ligands are of C–S type, with the combination of thiophene and NHC's, giving good soft coordination sites and are explored by Cavell and co-workers.78,79 Both of the works present good examples for the potential of sulphur in the coordination chemistry of coinage metal and provide a scope to develop other sulphur based orthogonal ligands, which can be further studied in terms of their coordination behaviour and photoluminescence properties. These examples show that many combinations in terms of hard soft interaction exists, which are not explored yet.
In the future, we expect a better understanding of the structure property relationship of these kind of compounds due to the increasing power of theoretical methods. This facilitates the task specific assembly of compounds showing a desired emission. Thus, on the long run, we expect that the emission colour can be adjusted by a rational synthesis, which is directed by theoretical predications.
Author contributions
VRN and FR did the literature research and wrote most parts of the manuscript. PWR originated the idea and supervised the work. All authors contributed to the preparation of the manuscript.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
We want to thank Niklas Reinfandt for proofreading the manuscript. The Deutsche Forschungsgemeinschaft (DFG) funded Research Training Group (RTG) 2039 (Molecular architecture for fluorescent cell imaging) is acknowledged for financial support of VRN. The DFG funded Transregional Collaborative Research Centre 88 [Cooperative Effects in Homo- and Heterometallic Complexes (3MET)], Projects C3, is acknowledged for financial support of FK.Notes and references
- V. W.-W. Yam, V. K.-M. Au and S. Y.-L. Leung, Chem. Rev., 2015, 115, 7589–7728 CrossRef CAS PubMed.
- S. Raju, H. B. Singh and R. J. Butcher, Dalton Trans., 2020, 49, 9099–9117 RSC.
- H. Schmidbaur and A. Schier, Chem. Soc. Rev., 2012, 41, 370–412 RSC.
- F. Scherbaum, A. Grohmann, G. Müller and H. Schmidbaur, Angew. Chem., Int. Ed. Engl., 1989, 28, 463–465 CrossRef.
- H. Schmidbaur, Gold Bull., 1990, 23, 11–21 CrossRef CAS.
- M. Jansen, Angew. Chem., Int. Ed. Engl., 1987, 26, 1098–1110 CrossRef.
- N. V. S. Harisomayajula, S. Makovetskyi and Y. C. Tsai, Chem. – Eur. J., 2019, 25, 8936–8954 CrossRef CAS PubMed.
- H. L. Hermann, G. Boche and P. Schwerdtfeger, Chem. – Eur. J., 2001, 7, 5333–5342 CrossRef CAS.
- H. Schmidbaur and A. Schier, Angew. Chem., Int. Ed., 2015, 54, 746–784 CrossRef CAS PubMed.
- M. Jansen, J. Less-Common Met., 1980, 76, 285–292 CrossRef CAS.
- J. M. López-de-Luzuriaga, M. Monge, M. E. Olmos, D. Pascual and M. Rodríguez-Castillo, Organometallics, 2012, 31, 3720–3729 CrossRef.
- J. M. López-De-Luzuriaga, M. Monge, M. E. Olmos, J. Quintana and M. Rodríguez-Castillo, Inorg. Chem., 2019, 58, 1501–1512 CrossRef PubMed.
- M. J. Calhorda, C. Ceamanos, O. Crespo, M. C. Gimeno, A. Laguna, C. Larraz, P. D. Vaz and M. D. Villacampa, Inorg. Chem., 2010, 49, 8255–8269 CrossRef CAS PubMed.
- A. Laguna, T. Lasanta, J. M. López-De-Luzuriaga, M. Monge, P. Naumov and M. E. Olmos, J. Am. Chem. Soc., 2010, 132, 456–457 CrossRef CAS PubMed.
- Q. Wan, J. Yang, W.-P. To and C.-M. Che, Proc. Natl. Acad. Sci. U. S. A., 2021, 118, e2019265118 CrossRef CAS PubMed.
- M. B. Brands, J. Nitsch and C. F. Guerra, Inorg. Chem., 2018, 57, 2603–2608 CrossRef CAS PubMed.
- S. Sculfort and P. Braunstein, Chem. Soc. Rev., 2011, 40, 2741–2760 RSC.
- S. Nayeri, S. Jamali, A. Jamjah and H. Samouei, Inorg. Chem., 2019, 58, 12122–12131 CrossRef CAS PubMed.
- Z. Lei, S.-S. Chang and Q.-M. Wang, Eur. J. Inorg. Chem., 2017, 5098–5102 CrossRef CAS.
- C. E. Strasser and V. J. Catalano, J. Am. Chem. Soc., 2010, 132, 10009–10011 CrossRef CAS PubMed.
- K. N. Jarzembska, R. Kamiński, K. F. Dziubek, M. Citroni, D. Paliwoda, K. Durka, S. Fanetti and R. Bini, Inorg. Chem., 2018, 57, 8509–8520 CrossRef CAS PubMed.
- K. Chen, M. M. Nenzel, T. M. Brown and V. J. Catalano, Inorg. Chem., 2015, 54, 6900–6909 CrossRef CAS PubMed.
- N. Glebko, T. M. Dau, A. S. Melnikov, E. V. Grachova, I. V. Solovyev, A. Belyaev, A. J. Karttunen and I. O. Koshevoy, Chem. – Eur. J., 2018, 24, 3021–3029 CrossRef CAS PubMed.
- X. L. Pei, Z. J. Guan, Z. A. Nan and Q. M. Wang, Angew. Chem., 2021, 133, 14502–14505 CrossRef.
- V. W.-W. Yam and K. K.-W. Lo, Chem. Soc. Rev., 1999, 28, 323–334 RSC.
- R. Jazzar, M. Soleilhavoup and G. Bertrand, Chem. Rev., 2020, 120, 4141–4168 CrossRef CAS PubMed.
- J. C. Y. Lin, R. T. W. Huang, C. S. Lee, A. Bhattacharyya, W. S. Hwang and I. J. B. Lin, Chem. Rev., 2009, 109, 3561–3598 CrossRef CAS PubMed.
- A. V. Paderina, I. O. Koshevoy and E. V. Grachova, Dalton Trans., 2021, 50, 6003–6033 RSC.
- V. W.-W. Yam and K. M.-C. Wong, Chem. Commun., 2011, 47, 11579 RSC.
- C. Fliedel, A. Ghisolfi and P. Braunstein, Chem. Rev., 2016, 116, 9237–9304 CrossRef CAS PubMed.
- A. Belyaev, T. M. Dau, J. Jänis, E. V. Grachova, S. P. Tunik and I. O. Koshevoy, Organometallics, 2016, 35, 3763–3774 CrossRef CAS.
- R. G. Pearson, J. Chem. Educ., 1968, 45, 643 CrossRef CAS.
- R. G. Pearson, Inorg. Chim. Acta, 1995, 240, 93–98 CrossRef CAS.
- W. H. Chan, K. K. Cheung, T. C. W. Mak and C. M. Che, J. Chem. Soc., Dalton Trans., 1998, 873–874 RSC.
- Y.-Y. Lin, S.-W. Lai, C.-M. Che, Fu, Z.-Y. Zhou and N. Zhu, Inorg. Chem., 2005, 44, 1511–1524 CrossRef CAS PubMed.
- A. Belyaev, T. Eskelinen, T. M. Dau, Y. Y. Ershova, S. P. Tunik, A. S. Melnikov, P. Hirva and I. O. Koshevoy, Chem. – Eur. J., 2018, 24, 1404–1415 CrossRef CAS PubMed.
- Z. Lei, X.-L. Pei, Z.-J. Guan and Q.-M. Wang, Angew. Chem., Int. Ed., 2017, 56, 7117–7120 CrossRef CAS PubMed.
- L.-Q. Mo, J.-H. Jia, L.-J. Sun and Q.-M. Wang, Chem. Commun., 2012, 48, 8691 RSC.
- X.-Y. Liu, Y. Yang, Z. Lei, Z.-J. Guan and Q.-M. Wang, Chem. Commun., 2016, 52, 8022–8025 RSC.
- S. Nayeri, S. Jamali, A. Jamjah, J. R. Shakirova, S. P. Tunik, V. Gurzhiy, H. Samouei and H. R. Shahsavari, Inorg. Chem., 2020, 59, 5702–5712 CrossRef CAS PubMed.
- C. Kaub, S. Lebedkin, A. Li, S. V. Kruppa, P. H. Strebert, M. M. Kappes, C. Riehn and P. W. Roesky, Chem. – Eur. J., 2018, 24, 6094–6104 CrossRef CAS PubMed.
- C. Kaub, S. Lebedkin, S. Bestgen, R. Köppe, M. M. Kappes and P. W. Roesky, Chem. Commun., 2017, 53, 9578–9581 RSC.
- T. Simler, K. Möbius, K. Müller, T. J. Feuerstein, M. T. Gamer, S. Lebedkin, M. M. Kappes and P. W. Roesky, Organometallics, 2019, 38, 3649–3661 CrossRef CAS.
- E. Fogler, E. Balaraman, Y. Ben-David, G. Leitus, L. J. W. Shimon and D. Milstein, Organometallics, 2011, 30, 3826–3833 CrossRef CAS.
- V. J. Catalano, M. A. Malwitz and A. O. Etogo, Inorg. Chem., 2004, 43, 5714–5724 CrossRef CAS.
- C. E. Strasser and V. J. Catalano, Inorg. Chem., 2011, 50, 11228–11234 CrossRef CAS PubMed.
- V. J. Catalano and A. L. Moore, Inorg. Chem., 2005, 44, 6558–6566 CrossRef CAS PubMed.
- S. Schäfer, M. T. Gamer, S. Lebedkin, F. Weigend, M. M. Kappes and P. W. Roesky, Chem. – Eur. J., 2017, 23, 12198–12209 CrossRef.
- S. Bestgen, M. T. Gamer, S. Lebedkin, M. M. Kappes and P. W. Roesky, Chem. – Eur. J., 2015, 21, 601–614 CrossRef CAS PubMed.
- V. J. Catalano and S. J. Horner, Inorg. Chem., 2003, 42, 8430–8438 CrossRef CAS PubMed.
- E. Hobbollahi, M. List, B. Hupp, F. Mohr, R. J. F. Berger, A. Steffen and U. Monkowius, Dalton Trans., 2017, 46, 3438–3442 RSC.
- K. N. Jarzembska, R. Kamiński, B. Fournier, E. Trzop, J. D. Sokolow, R. Henning, Y. Chen and P. Coppens, Inorg. Chem., 2014, 53, 10594–10601 CrossRef CAS PubMed.
- I. O. Koshevoy, J. R. Shakirova, A. S. Melnikov, M. Haukka, S. P. Tunik and T. A. Pakkanen, Dalton Trans., 2011, 40, 7927 RSC.
- V. J. Catalano, J. M. López-De-Luzuriaga, M. Monge, M. E. Olmos and D. Pascual, Dalton Trans., 2014, 43, 16486–16497 RSC.
- K. Chen and V. J. Catalano, Eur. J. Inorg. Chem., 2015, 5254–5261 CAS.
- N. Tsukada, O. Tamura and Y. Inoue, Organometallics, 2002, 21, 2521–2528 CrossRef CAS.
- M. Dahlen, E. H. Hollesen, M. Kehry, M. T. Gamer, S. Lebedkin, D. Schooss, M. M. Kappes, W. Klopper and P. W. Roesky, Angew. Chem., Int. Ed., 2021, 60, 23365–23372 CrossRef CAS PubMed.
- M. Dahlen, M. Kehry, S. Lebedkin, M. M. Kappes, W. Klopper and P. W. Roesky, Dalton Trans., 2021, 50, 13412–13420 RSC.
- H. Schmidbaur and A. Schier, Chem. Soc. Rev., 2012, 41, 370–412 RSC.
- G. A. Bowmaker, Effendy, S. Marfuah, B. W. Skelton and A. H. White, Inorg. Chim. Acta, 2005, 358, 4371–4388 CrossRef.
- M. T. Dau, J. R. Shakirova, A. J. Karttunen, E. V. Grachova, S. P. Tunik, A. S. Melnikov, T. A. Pakkanen and I. O. Koshevoy, Inorg. Chem., 2014, 53, 4705–4715 CrossRef CAS PubMed.
- P. Ai, A. A. Danopoulos, P. Braunstein and K. Y. Monakhov, Chem. Commun., 2014, 50, 103–105 RSC.
- F. A. Cotton, X. Feng, M. Matusz and R. Poli, J. Am. Chem. Soc., 1988, 110, 7077–7083 CrossRef CAS.
- A. C. Lane, M. V. Vollmer, C. H. Laber, D. Y. Melgarejo, G. M. Chiarella, J. P. Fackler, X. Yang, G. A. Baker and J. R. Walensky, Inorg. Chem., 2014, 53, 11357–11366 CrossRef CAS PubMed.
- H. E. Abdou, A. A. Mohamed and J. P. Fackler, Inorg. Chem., 2005, 44, 166–168 CrossRef CAS PubMed.
- D. Fenske, G. Baum, A. Zinn and K. Dehnicke, Z. Naturforsch., B: J. Chem. Sci., 1990, 45, 1273–1278 CrossRef CAS.
- F. H. Allen, O. Kennard, D. G. Watson, L. Brammer, A. G. Orpen and R. Taylor, J. Chem. Soc., Perkin Trans. 2, 1987, S1–S19 RSC.
- M. Dahlen, T. P. Seifert, S. Lebedkin, M. T. Gamer, M. M. Kappes and P. W. Roesky, Chem. Commun., 2021, 57, 13146–13149 RSC.
- E. Hartmann and J. Strähle, Z. Naturforsch., B: J. Chem. Sci., 1989, 44b, 1–4 Search PubMed.
- P. N. Bartlett, F. Cheng, D. A. Cook, A. L. Hector, W. Levason, G. Reid and W. Zhang, Inorg. Chim. Acta, 2010, 363, 1048–1051 CrossRef CAS.
- M. Stollenz, Chem. – Eur. J., 2019, 25, 4274–4298 CrossRef CAS PubMed.
- H. de la Riva, M. Nieuwhuyzen, C. Mendicute Fierro, P. R. Raithby, L. Male and M. C. Lagunas, Inorg. Chem., 2006, 45, 1418–1420 CrossRef CAS PubMed.
- M. Gil-Moles, M. C. Gimeno, J. M. López-de-Luzuriaga, M. Monge, M. E. Olmos and D. Pascual, Inorg. Chem., 2017, 56, 9281–9290 CrossRef CAS PubMed.
- O. Crespo, E. J. Fernández, M. Gil, M. Concepción Gimeno, P. G. Jones, A. Laguna, J. M. López-De-Luzuriaga and M. Elena Olmos, J. Chem. Soc., Dalton Trans., 2002, 1319–1326 RSC.
- T. P. Seifert, S. Bestgen, T. J. Feuerstein, S. Lebedkin, F. Krämer, C. Fengler, M. T. Gamer, M. M. Kappes and P. W. Roesky, Dalton Trans., 2019, 48, 15427–15434 RSC.
- T. M. Dau, B. D. Asamoah, A. Belyaev, G. Chakkaradhari, P. Hirva, J. Jänis, E. V. Grachova, S. P. Tunik and I. O. Koshevoy, Dalton Trans., 2016, 45, 14160–14173 RSC.
- Y. Takino, N. Yoshinari, T. Kawamoto and T. Konno, Chem. Lett., 2012, 41, 834–836 CrossRef CAS.
- D. J. Nielsen, K. J. Cavell, M. S. Viciu, S. P. Nolan, B. W. Skelton and A. H. White, J. Organomet. Chem., 2005, 690, 6133–6142 CrossRef CAS.
- M. Bierenstiel and E. D. Cross, Coord. Chem. Rev., 2011, 255, 574–590 CrossRef CAS.
Footnote |
| † These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2022 |