
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Unearthing the unique stability of thiophosphonium-C-terminal cysteine adducts on peptides and proteins†
Richard J.
Spears
 ,
Alina
Chrzastek
,
Steven Y.
Yap
,
Kersti
Karu
,
Abil E.
Aliev
,
James R.
Baker
* and
Vijay
Chudasama
*
,
Alina
Chrzastek
,
Steven Y.
Yap
,
Kersti
Karu
,
Abil E.
Aliev
,
James R.
Baker
* and
Vijay
Chudasama
*
UCL Department of Chemistry, 20 Gordon Street, London WC1H 0AJ, UK. E-mail: v.chudasama@ucl.ac.uk; j.r.baker@ucl.ac.uk
First published on 2nd April 2022
Abstract
Herein we report a fundamental discovery on the use of tris(dialkylamino)phosphine reagents for peptide and protein modification. We discovered that C-terminal thiophosphonium species, which are uniquely stable, could be selectively and rapidly generated from their disulfide counterparts. In sharp and direct contrast, internal thiophosphonium species rapidly degrade to dehydroalanine. We demonstrate this remarkable chemoselectivity on a bis-cysteine model peptide, and the formation of a stable C-terminal-thiophosphonium adduct on an antibody fragment, as well as characterise the species in various small molecule/peptide studies.
Protein modification has established itself as a cornerstone of chemical biology over the past two decades.1,2 Applications of this field are wide ranging; examples include design of imaging agents3 and novel therapeutics such as antibody–drug conjugates (ADCs).4 Typically, protein modification occurs at an amino acid side chain, such as lysine5 or tyrosine.6,7 Cysteine in particular is regularly used due to the suitability of its thiol side chain8–11 (ease of incorporation via conventional site-directed mutagenesis, high nucleophilicity at neutral pH, low natural abundance which assists in avoiding heterogeneity).12 In addition to side chain modification, the N-terminus is frequently used for site-specific modification due to the unique properties of the α-amino group compared to those of the ε-amino groups of lysine residues (e.g. pKa 6–8 vs. pKaca. 10).13 Occasionally, strategies targeting the N-terminus use both the α-amino group and amino acid side chain, as seen for N-terminal cysteine modification using cyanobenzothiazole derivatives.14,15 In contrast to the N-terminus or side chains, examples of chemical modification specific to the C-terminus are comparatively rare; enzymatic-based strategies tend to dominate this area of research.16 This is logical given the challenge of both activating and selectively targeting the carboxylic acid group of the C-terminus in the presence of glutamate and aspartate carboxylic acid side chains. Currently, the only two examples reported in the literature of chemical bioconjugation specific for proteins bearing canonical amino acid residues at the C-terminus include (i) an N → S acyl transfer of C-terminal cysteines to generate thioesters, which can be converted to hydrazides using subsequent native chemical ligation (NCL)17 and (ii) photoredox-mediated decarboxylative addition of Michael acceptors to the C-terminus, which selectively targets the C-terminus carboxylate over glutamate/aspartic acid carboxylates through differences in oxidative potentials.18 We therefore hypothesised that investigating protein modification strategies focused on the C-terminus would be of high value. In particular, we envisaged that potential modifications specifically on the C-terminus side chain could be “locked” into place by the adjacent carboxylic acid (akin to that of the α-amino group of the N-terminus for N-terminal modifications). We decided to focus on C-terminal cysteine, given cysteine's aforementioned favourable properties, anticipating that a modification strategy for a C-terminal cysteine may have a different outcome compared to modification of cysteine residues at internal positions within an amino acid sequence. Moreover, several peptides and proteins (e.g. the majority of therapeutic full antibodies)19 contain a natural C-terminal cysteine residue.
During our investigation of chemical modification strategies focused at the C-terminus, our attention was turned towards tris(dialkylamino)phosphines. These reagents, specifically tris(dimethylamino)phosphine/hexamethylphosphorous triamide, (HMPT 1), have previously been reported for use in activation of alkyl thiols towards nucleophilic displacement reactions in organic synthesis,20 desulfurization reactions21,22 and in formation of dehydroalanine (Dha) on small molecules and proteins.23 All of the aforementioned transformations proceed via a thiophosphonium intermediate, which are traditionally regarded as highly reactive and difficult to isolate.24 In some specific cases, however, the thiophosphonium intermediate has been isolated either as a small molecule salt20 or stabilised through use of rotaxanes,24 or observed by LCMS upon treating a disulfide containing protein with HMPT 1.23 We were particularly intrigued how the thiophosphonium motif might behave when located at the C-terminus near the carboxylate group, and whether or not the presence of a nearby carboxylate may (i) impart stabilising interactions between the thiophosphonium cation and carboxylate, (ii) affect propensity of thiophosphonium species towards nucleophilic attack/desulfurisation, and (iii) affect the pKa of the cysteine α proton, and therefore affect the likelihood of the thiophosphonium decomposition to give Dha.
To begin with, we reacted N-acetyl cysteine 2 with 4-nitrophenyl disulfide 3 to synthesise model N-acetyl cysteine disulfide 4 for use in validation of thiophosphonium formation. We chose to use unsymmetrical alkyl–aryl disulfides (S–SAr) as these have previously been reported to assist in facilitating regioselective phosphine attack at the alkyl sulfur of the S–SAr disulfide,25 generating the desired thiophosphonium species. We initially screened a panel of commercially available phosphine reagents 1 and 5–11 against disulfide 4 in MeOH, and analysed formation of thiophosphonium by LCMS analysis (Fig. 1a). Tris(dialkylamino)phosphines 1, 5, 6, and 7 all gave rise to thiophosphonium cations 12–15 (respectively) as judged by LCMS. Minor thiophosphonium formation was noted in the case of mixed N-/O-phosphine 8 to give 16, whereas no thiophosphonium species 17–19 was detected when using mixed N-/O-phosphine 9 and alkyl phosphines tris(2-carboxyethyl)phosphine (TCEP) 10 and tris(hydroxypropyl)phosphine (THPP) 11. We next sought to see if one of these thiophosphonium species could both be formed in aqueous conditions and isolated for characterisation. We chose thiophoshonium 12 for this purpose, as HMPT 1 has been reported before for formation of thiophosphonium intermediates. We found that thiophosphonium 12 could additionally be formed under aqueous conditions in H2O![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :MeCN (1:1) as judged by LCMS and subsequently purified by semi-preparative high-pressure liquid chromatography (HPLC) in a 52% yield. 31P NMR of 12 revealed a single peak at δ 65.6 ppm, which was in accordance with previously reported 31P NMR data for thiophosphonium salts derived from HMPT 1.26 Thiophosphonium 12 formation was further confirmed via tandem mass spectrometry (MSMS), with a fragment corresponding to the thiophosphonium motif as the major species following MS1 (Fig. 1b). Thiophosphonium 12 proved resistant to decomposition when incubated in 0.2 M PB pH 8 buffer for 18 h and, in buffer, proved inert towards nucleophiles previously reported to facilitate nucleophilic displacement of the thiophosphonium group (see ESI†).20 Thiophosphonium 12 also proved to be completely resistant to strongly acidic conditions (10 mM HCl, 18 h, 37 °C), with only partial decomposition of the thiophosphonium motif observed under strongly basic conditions (10 mM NaOH, 18 h, 37 °C, see ESI†).
:MeCN (1:1) as judged by LCMS and subsequently purified by semi-preparative high-pressure liquid chromatography (HPLC) in a 52% yield. 31P NMR of 12 revealed a single peak at δ 65.6 ppm, which was in accordance with previously reported 31P NMR data for thiophosphonium salts derived from HMPT 1.26 Thiophosphonium 12 formation was further confirmed via tandem mass spectrometry (MSMS), with a fragment corresponding to the thiophosphonium motif as the major species following MS1 (Fig. 1b). Thiophosphonium 12 proved resistant to decomposition when incubated in 0.2 M PB pH 8 buffer for 18 h and, in buffer, proved inert towards nucleophiles previously reported to facilitate nucleophilic displacement of the thiophosphonium group (see ESI†).20 Thiophosphonium 12 also proved to be completely resistant to strongly acidic conditions (10 mM HCl, 18 h, 37 °C), with only partial decomposition of the thiophosphonium motif observed under strongly basic conditions (10 mM NaOH, 18 h, 37 °C, see ESI†).
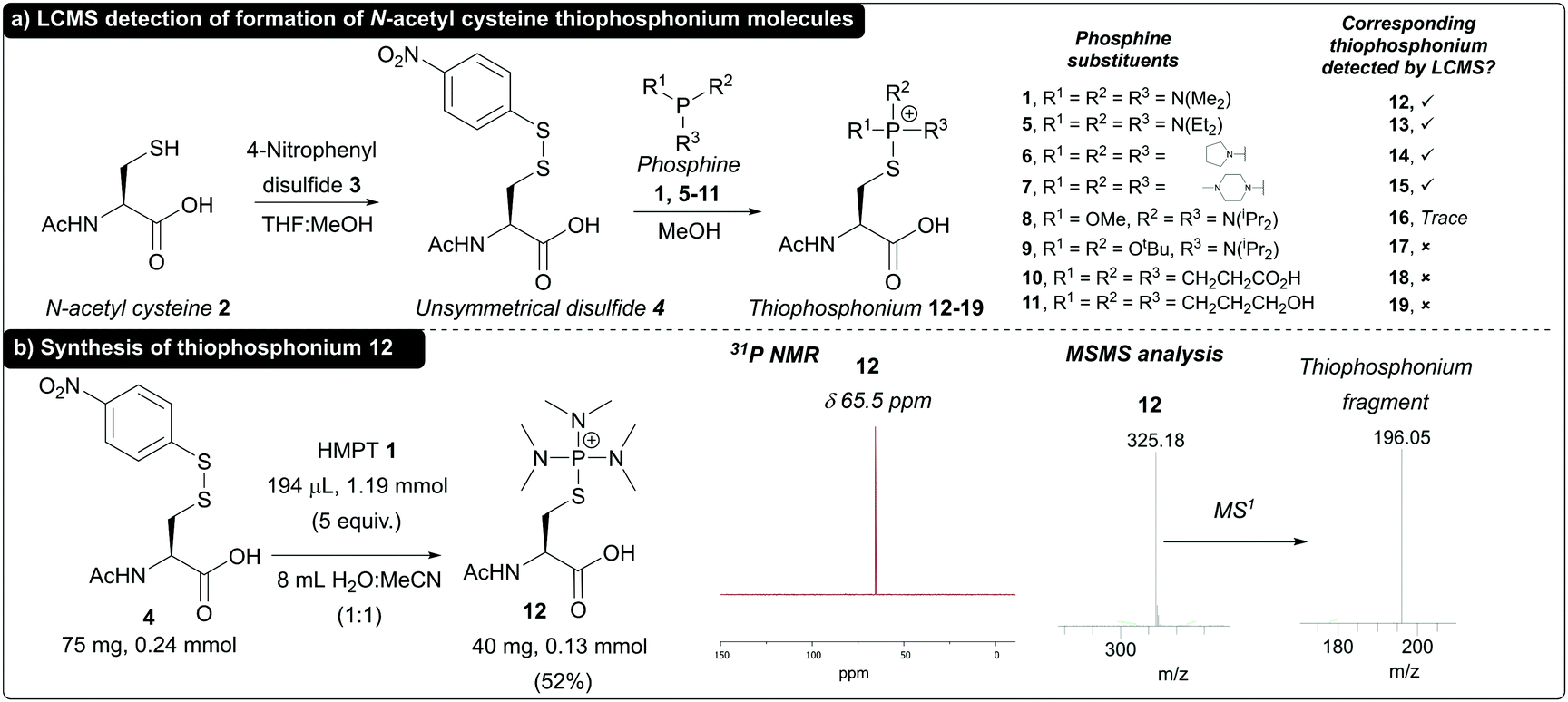 | ||
| Fig. 1 (a) Formation of thiophosphonium species and detection by LCMS (b) synthesis of thiophosphonium 12 and 31P NMR and MSMS data for isolated 12. | ||
Next, thiophosphonium formation on small peptide units was investigated using HMPT 1. Peptides FEKGC 20, FCEKG 21 and CFEKG 22 (containing cysteine at the C-terminus, at an internal position and at the N-terminus respectively) were synthesised via solid phase peptide synthesis (SPPS) and subsequently derivatised with Ellman's reagent 23 to give the corresponding disulfide-containing peptides 24 (C-terminal), 25 (internal) and 26 (N-terminal, Fig. 2, see ESI† for full details). To begin with, peptide 24 was subjected to HMPT 1 in PB pH 8.0 (10% MeCN) at 37 °C, and the results were analysed by LCMS. Full conversion to thiophosphonium peptide species 27 (Fig. 3a) was clearly observed at t = 5 min (as judged by LCMS, see ESI†). We additionally saw formation of a previously reported SAr thiophosphonium species (see ESI†).23 Thiophosphonium species 27 exhibited stability during incubation at 37 °C at t = 1 h and t = 18 h (see ESI†) and MSMS analysis further confirmed the thiophosphonium as being located on the C-terminus. We next treated peptide 25 with HMPT 1. Initially, this resulted in thiophosphonium 28 formation at t = 5 min (see ESI†); in contrast to thiophosphonium 27, however, thiophosphonium 28 had almost completely degraded at t = 1 h (as observed by LCMS, see ESI†). We hypothesised, in accordance with previous literature, decomposition of the thiophosphonium species to Dha to give peptide 29 was occurring following HMPT 1 addition (Fig. 3b). Although Dha peptide 29 could not be reliably observed in LCMS analysis, Dha formation was confirmed through thiol addition of n-hexanethiol 30 to the reaction mixture of peptide 28 and HMPT 1 to give thiol-capped peptide 31, suggesting Dha 29 as the intermediate resulting from thiophosphonium degradation (see ESI†). Finally, we treated peptide 26 with HMPT 1. Thiophosphonium 32 formation was observed, with some degradation after 18 h of incubation observed via LCMS (see ESI†). We observed a peak in the LCMS trace which could have corresponded to the associated Dha peptide or ketone-containing peptide 33, which could have formed through degradation of thiophosphonium 32 to the corresponding Dha/enamine-containing peptide, followed by tautomerisation to an imine and finally hydrolysis to the corresponding ketone 33. Addition of n-hexanethiol 30 to the reaction mixture gave no reaction as observed via LCMS, whilst addition of benzyloxyamine 34 under slightly acidic conditions gave a new peak in the LCMS trace corresponding to oxime product 35 (see ESI†). We thus hypothesised that the peak corresponded to ketone-containing peptide 33.
 | ||
| Fig. 2 Cysteine-containing peptides and their disulfide-containing counterparts. | ||
 | ||
| Fig. 3 (a) Reaction of peptide 24 with HMPT 1 to give the stable C-terminal thiophosphonium 27 as confirmed by LCMS and MSMS analysis. (b) Reaction of peptide 25 with HMPT 1 to give internal thiophosphonium 28, which rapidly degrades to Dha 29, which can be modified with n-hexanethiol 30 to give peptide 31. (c) Reaction of peptide 26 with HMPT 1 to give N-terminal thiophosphonium 32 as a major product and ketone 33 as a minor product. Ketone 33 can be functionalised with aminooxy 34 to give oxime product 35. | ||
To further demonstrate this difference in reactivity, we next turned our attention towards the model peptide FCEKAGC 36, which contains two cysteine residues; Cys2 at an internal position and Cys7 at the C-terminus of the peptide. Following derivatisation of the thiol groups with Ellman's reagent 23 to give peptide 37, the peptide was then treated with HMPT 1 to generate Dha at position 2 and a thiophosphonium adduct at position 7 (peptide 38). Further treatment with n-hexanethiol 30 subsequently gave dually modified peptide 39 (Fig. 4a), which was isolated as two diastereoisomers by preparative-HPLC and confirmed by LCMS and MSMS analysis (Fig. 4b).
 | ||
| Fig. 4 (a) Dual modification of peptide 36 to give dually modified peptide 39, with confirmation by LCMS analysis and purification by HPLC. (b) MSMS analysis of 39. | ||
Finally, as a proof of concept we tested HMPT 1 within the context of modifying a protein containing a C-terminal cysteine. Specifically, we focused on modifying the C-terminal cysteine of the light chain of the herceptin (Trastuzumab) Fab fragment. Initially, reduction of the Fab disulfide bond and capping of the majority of the heavy chain free thiol only was performed (see ESI† for details), i.e. to give the free thiol light chain and thiol-capped heavy chain as the major products. The Fab light chain free thiol was then derivatised with Ellman's reagent to give the corresponding light chain aryl thiol disulfide. This was subsequently treated with HMPT 1, which generated the thiophosphonium species with complete conversion from the Elman's capped light chain. LCMS analysis confirmed the presence of the Fab light chain thiophosphonium, with no decomposition to Dha observed (see ESI† for details).
To conclude, we have demonstrated the synthesis of a thiophosphonium species based on N-acetyl cysteine, along with C-terminal cysteine-containing peptides and trastuzumab Fab light chain. These thiophosphonium species displayed unexpected stability and could be subsequently isolated and analysed by NMR and/or MS. In contrast, thiophosphonium formation at a cysteine positioned internally within a peptide sequence resulted in rapid decomposition to Dha. We hypothesise that the presence of an adjacent carboxylic acid imparts stabilising interactions that prevent decomposition of the C-terminal thiophosphonium, either through ionic interactions between the carboxylic acid and thiophosphonium, or through a reduced propensity for abstraction of the alpha proton and subsequent collapse of the thiophosphonium species to Dha. We intend to explore this difference in reactivity of C-terminal and internal cysteines for further development of protein modification strategies in future work, but share our current findings here in view of the high impact of this fundamental discovery.
We gratefully acknowledge the Leverhulme Trust (RPG-2017-288, RPG-2020-010) and EPSRC (EP/T016043/1) for funding.
Conflicts of interest
V. C. and J. R. B. are Directors of the spin-out ThioLogics, but there are no competing financial interests to declare.References
- C. D. Spicer and B. G. Davis, Nat. Commun., 2014, 5, 4740 CrossRef CAS PubMed.
- E. A. Hoyt, P. M. S. D. Cal, B. L. Oliveira and G. J. L. Bernardes, Nat. Rev. Chem., 2019, 3, 147–171 CrossRef CAS.
- P. Agarwal, B. J. Beahm, P. Shieh and C. R. Bertozzi, Angew. Chem., Int. Ed., 2015, 54, 11504–11510 CrossRef CAS PubMed.
- S. J. Walsh, J. D. Bargh, F. M. Dannheim, A. R. Hanby, H. Seki, A. J. Counsell, X. Ou, E. Fowler, N. Ashman, Y. Takada, A. Isidro-Llobet, J. S. Parker, J. S. Carroll and D. R. Spring, Chem. Soc. Rev., 2021, 50, 1305–1353 RSC.
- M. J. Matos, B. L. Oliveira, N. Martínez-Sáez, A. Guerreiro, P. M. S. D. S. D. Cal, J. Bertoldo, M. Maneiro, E. Perkins, J. Howard, M. J. Deery, J. M. Chalker, F. Corzana, G. Jiménez-Osés and G. J. L. L. Bernardes, J. Am. Chem. Soc., 2018, 140, 4004–4017 CrossRef CAS PubMed.
- D. Alvarez Dorta, D. Deniaud, M. Mével and S. G. Gouin, Chem. – Eur. J., 2020, 26, 14257–14269 CrossRef CAS PubMed.
- P. A. Szijj, K. A. Kostadinova, R. J. Spears and V. Chudasama, Org. Biomol. Chem., 2020, 18, 9018–9028 RSC.
- C. Zhang, M. Welborn, T. Zhu, N. J. Yang, M. S. Santos, T. Van Voorhis and B. L. Pentelute, Nat. Chem., 2016, 8, 120–128 CrossRef CAS PubMed.
- C. Bahou, D. A. Richards, A. Maruani, E. A. Love, F. Javaid, S. Caddick, J. R. Baker and V. Chudasama, Org. Biomol. Chem., 2018, 16, 1359–1366 RSC.
- M. A. Kasper, M. Glanz, A. Stengl, M. Penkert, S. Klenk, T. Sauer, D. Schumacher, J. Helma, E. Krause, M. C. Cardoso, H. Leonhardt and C. P. R. Hackenberger, Angew. Chem., Int. Ed., 2019, 58, 11625–11630 CrossRef CAS PubMed.
- E. Gil de Montes, A. Istrate, C. D. Navo, E. Jiménez-Moreno, E. A. Hoyt, F. Corzana, I. Robina, G. Jiménez-Osés, A. J. Moreno-Vargas and G. J. L. Bernardes, Angew. Chem., Int. Ed., 2020, 59, 6196–6200 CrossRef CAS PubMed.
- O. Koniev and A. Wagner, Chem. Soc. Rev., 2015, 44, 5495–5551 RSC.
- C. B. Rosen and M. B. Francis, Nat. Chem. Biol., 2017, 13, 697–705 CrossRef CAS PubMed.
- L. Cui and J. Rao, Methods Mol. Biol., 2015, 1266, 81–92 CrossRef CAS PubMed.
- H. Ren, F. Xiao, K. Zhan, Y. P. Kim, H. Xie, Z. Xia and J. Rao, Angew. Chem., Int. Ed., 2009, 48, 9658–9662 CrossRef CAS PubMed.
- Y. Zhang, K. Y. Park, K. F. Suazo and M. D. Distefano, Chem. Soc. Rev., 2018, 47, 9106–9136 RSC.
- A. L. Adams, B. Cowper, R. E. Morgan, B. Premdjee, S. Caddick and D. Macmillan, Angew. Chem., Int. Ed., 2013, 52, 13062–13066 CrossRef CAS PubMed.
- S. Bloom, C. Liu, D. K. Kölmel, J. X. Qiao, Y. Zhang, M. A. Poss, W. R. Ewing and D. W. C. C. MacMillan, Nat. Chem., 2018, 10, 205–211 CrossRef CAS PubMed.
- Y. Shen, L. Zeng, A. Zhu, T. Blanc, D. Patel, A. Pennello, A. Bari, S. Ng, K. Persaud, Y. K. Kang, P. Balderes, D. Surguladze, S. Hindi, Q. Zhou, D. L. Ludwig and M. Snavely, MAbs, 2013, 5, 418–431 CrossRef PubMed.
- G. A. Krafft and T. L. Siddall, Tetrahedron Lett., 1985, 26, 4867–4870 CrossRef CAS.
- G. J. L. Bernardes, E. J. Grayson, S. Thompson, J. M. Chalker, J. C. Errey, F. El Oualid, T. D. W. Claridge and B. G. Davis, Angew. Chem., Int. Ed., 2008, 47, 2244–2247 CrossRef CAS PubMed.
- M. S. Collins, M. E. Carnes, B. P. Nell, L. N. Zakharov and D. W. Johnson, Nat. Commun., 2016, 7, 11052 CrossRef CAS PubMed.
- J. M. Chalker, S. B. Gunnoo, O. Boutureira, S. C. Gerstberger, M. Fernández-González, G. J. L. Bernardes, L. Griffin, H. Hailu, C. J. Schofield and B. G. Davis, Chem. Sci., 2011, 2, 1666–1676 RSC.
- T. Oku, Y. Furusho and T. Takata, Org. Lett., 2003, 5, 4923–4925 CrossRef CAS PubMed.
- D. N. Harpp and J. G. Gleason, J. Org. Chem., 1971, 36, 73–80 CrossRef CAS PubMed.
- R. K. Haynes and C. Indorato, Aust. J. Chem., 1984, 37, 1183–1194 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See https://doi.org/10.1039/d2cc01090a |
| This journal is © The Royal Society of Chemistry 2022 |