
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Tethered photocatalyst-directed palladium-catalysed C–H allenylation of N-aryl tetrahydroisoquinolines†
Mingfeng
Li
,
Xiu Li
Chia
and
Ye
Zhu
 *
*
Department of Chemistry, Faculty of Science, National University of Singapore, 3 Science Drive 3, 117543, Singapore. E-mail: chmzhu@nus.edu.sg
First published on 9th March 2022
Abstract
Harnessing radical intermediates in regioselective reactions presents a substantial challenge. Here, we report a novel control strategy through engineering covalently tethered transition metal–photocatalysts that conjoin Pd–phosphine and Ru/Ir photoredox units. This strategy allows us to override the innate regioselectivity of the Pd-catalysed C–H allenylation of N-aryl tetrahydroisoquinolines.
Nature has evolved a myriad of enzymes capable of generating highly reactive radical intermediates for challenging chemical transformations.1 Besides triggering a single electron transfer cascade, the active site of a radical enzyme orients the transient radical towards a specific part of a substrate through exquisite preorganization to achieve accurate selectivity and avoid damage to its own interior.2 Bifunctional photocatalysts capable of promoting synergistic radical generation and bond formation can exert such proximity and orientation effects to engender selectivity that is unattainable using dual catalysts. Since the seminal work from Bach and co-workers on the organocatalytic, photoinduced single electron transfer directed by hydrogen bonding interactions,3 this strategy has gained much traction through the design of ingenious photocatalysts incorporating aminocatalysts,4 Brønsted acids,5 and Lewis acids.6,7
During recent years, bifunctional transition metal complexes that promote both photoinduced single electron transfer and cross-coupling reactions have emerged as powerful tools in the synthetic chemists’ arsenal (Scheme 1A, a).8–10 By contrast, the strategy of covalently tethering a designated photocatalyst to a transition metal catalyst has remained unexploited in harnessing radical intermediates in cross-coupling reactions (Scheme 1A, b),11 despite the tremendous advances in the field of dual photoredox/transition metal catalysis.12,13 Conceivably, the approach of merging the diversity of photocatalysts with a variety of cross-coupling catalysts via covalent linkages could significantly expand the pool of transition metal photocatalysts. Furthermore, it presents an untapped opportunity for selectivity control over catalytic processes involving transient radical intermediates through exploiting the concomitant proximity and orientation effects.
 | ||
| Scheme 1 Tethered transition metal–photocatalysts enable a novel selectivity controlling strategy of C–H allenylation reactions. LG: leaving group; SET: single electron transfer. | ||
In pursue of such a strategy, we explored a Pd-catalysed C–C bond formation between propargylic alcohol derivatives and photoinduced radicals. We were intrigued by the diverse regioselectivity of the Pd-catalysed reactions of the propargylic alcohol derivatives. Depending on the type of nucleophilic coupling partner, reactions at all three carbon atoms are achievable, affording functionalised allenes, alkynes, and alkenes as the products, respectively (Scheme 1B, a).14 In a significant advance, Chen and Liang developed a dual photoredox/Pd-catalysed propargylic substitution using benzyl 1,4-dihydropyridine derivatives as radical precursors (Scheme 1B, b).15 The alkynyl products were obtained exclusively, irrespective of the phosphine ligand of the Pd catalyst. To date, the applications of radical precursors via C–H cleavage in the Pd-catalysed reactions of propargylic alcohol derivatives have not been reported. Importantly, regioselectivity control of such processes, particularly overriding the innate preferences for alkynylation, has not been achieved.16 Herein, we report the development of a tethered photocatalyst–Pd-catalysed C–H allenylation of N-aryl tetrahydroisoquinolines using propargylic acetate derivatives as coupling partners (Scheme 1C).
We commenced our study by evaluating the viability of cross-coupling between N-phenyl tetrahydroisoquinoline (1) and propargylic acetate derivative (2) using photoredox/Pd co-catalysts. Irradiated with blue LEDs, the reaction using an Ir(dtbbpy)(ppy)2PF6/Pd(PPh3)4 dual catalyst failed to yield the desirable products despite the consumption of substrates (Scheme 2a). This result indicates that the reaction under investigation differs from allylation17 of 1, which is known to proceed in the presence of the dual catalyst.17a In an effort to identify suitable catalysts, we investigated various combinations of phosphine ligands and photocatalysts. Gratifyingly, the reaction using Ru(bpy)3(PF6)2 as the photocatalyst and SPhos as a ligand for Pd produced coupling products under blue LED irradiation, albeit as a mixture of allene (3, 38%) and alkyne (4, 40%) (Scheme 2b). The formation of the alkynylation product is not unexpected. Chen and Liang have shown that alkynylation was observed when a dialkylbiphenyl phosphine (i.e., DavePhos) was utilised (Scheme 1B, b).15 Additionally, the formation of mixed products suggests that the putative α-amino radical intermediate18 likely reacts through a different mechanism than typical nucleophiles. Ma and co-workers have shown that Pd–SPhos-catalysed Negishi reactions of propargylic carbonates yield allenes exclusively.19
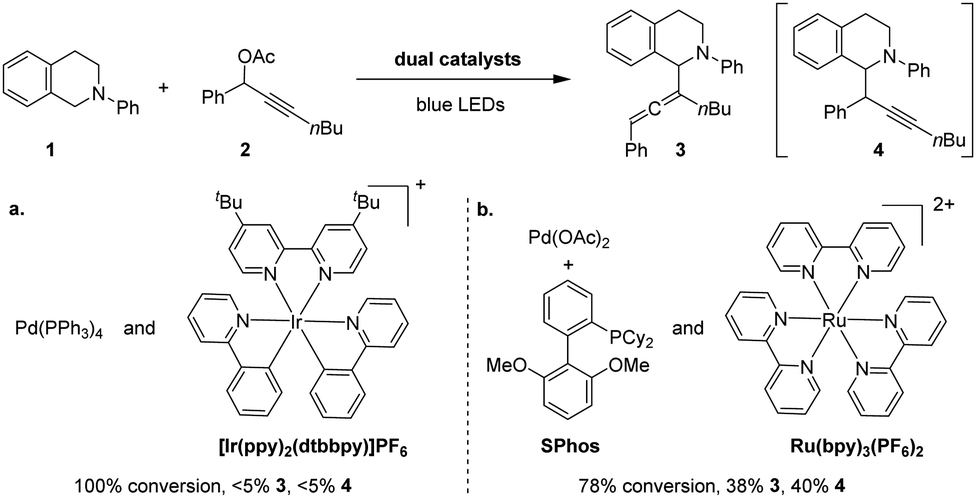 | ||
Scheme 2 C–H allenylation/alkynylation reactions using dual catalysts. Reaction conditions: (a) 1 (0.1 mmol), 2 (0.12 mmol), Pd(PPh3)4 (5.0 mol%), Ir(ppy)2(dtbbpy)PF6 (2.0 mol%), CH3CN (0.1 M), 19 W blue LEDs for 48 h. (b) 1 (0.1 mmol), 2 (0.2 mmol), Pd(OAc)2 (2.0 mol%), SPhos (1.0 mol%), Ru(bpy)3(PF6)2 (1.0 mol%), 2,6-lutidine (0.2 mmol), CH3CN (0.1 M), 19 W blue LEDs for 7 h. Conversions and yields were determined by the 1H NMR spectra using dibromomethane as an internal standard; 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 dr. :1 dr. | ||
Encouraged by the high reactivity of the Ru(bpy)3(PF6)2/Pd–SPhos dual catalyst system, we next devised a tethered transition metal photocatalyst to tackle the challenges arising from the regioselectivity control involving radical intermediates. Studies using mass spectrometry confirmed the existence of an adduct of 2 and Pd–SPhos as the presumptive intermediate prior to the C–C bond formation (Fig. S2 in the ESI†). Conceivably, the spatial dispositions of α-amino radicals and the allenyl/propargyl Pd–SPhos complex play a significant role in modulating the regioselectivity of the coupling step. We have recently achieved stereocontrol by a bifunctional Pd catalyst built on a dialkylbiphenyl phosphine scaffold.20 Following this principle, the preorganisation of a photocatalyst and Pd complex through a covalent linkage could orient the transient radical intermediate towards a specific site of the allenyl/propargyl Pd–SPhos intermediate (Scheme 1C).
To explore this strategy for regioselectivity control, we synthesised three covalently tethered transition metal photocatalysts (Scheme 3A). A 2,2′-bipyridine moiety was installed at the 3′-position of SPhos in three steps. Subsequent complexation with Ru and Ir precursors yielded the corresponding tethered SPhos-photocatalysts (SPhos-PC1–3). The covalent linkage does not alter the absorption and emission spectra of the photocatalyst in the visible light range (see ESI†).
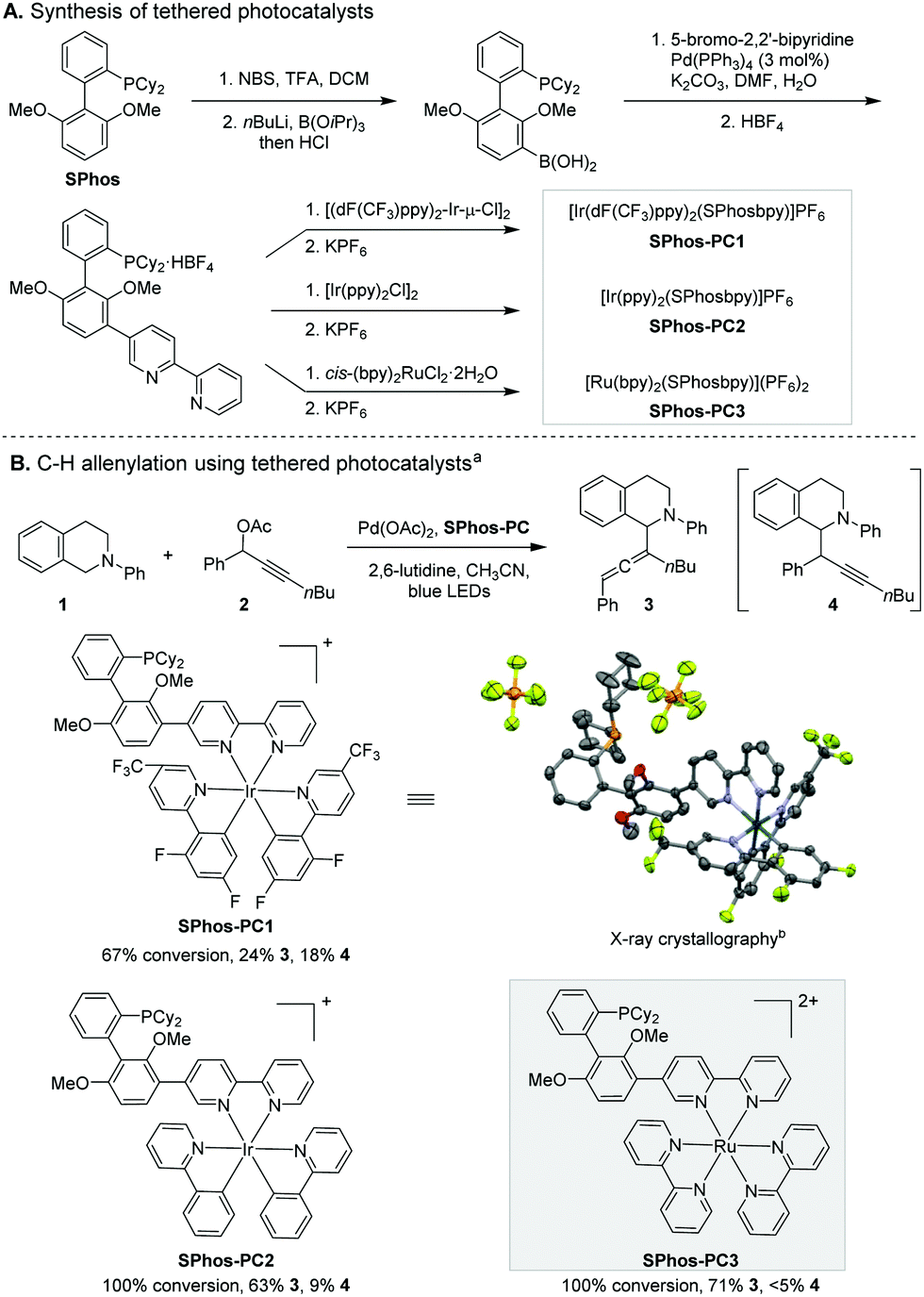 | ||
| Scheme 3 C–H allenylation reaction using novel tethered photocatalysts. aReaction conditions: 1 (0.1 mmol), 2 (0.2 mmol), Pd(OAc)2 (2.0 mol%), SPhos-PC (1.0 mol%), 2,6-lutidine (0.2 mmol), CH3CN (0.1 M), 19 W blue LEDs for 7 h. bH atoms have been omitted for clarity. Displacement ellipsoids are depicted at the 50% probability level. | ||
The reaction using SPhos-PC1 favoured the allenylation product 3 slightly (Scheme 3B). The steric bulk of the photocatalyst alone does not confer high selectivity. To our surprise, the use of another Ir-based SPhos-PC2 resulted in a marked increase in regioselectivity (3:4 ratio = 7:1). Apart from their different oxidation potentials, the dynamic steric effects of the photocatalyst moieties (i.e., CF3 substituents21 of SPhos-PC1) could induce conformational changes critical for regioselectivity control. Recent computational studies have revealed the localization of electron density at the bipyridine ligand of heteroleptic [Ir(ppy)2(bpy)]+ and the high delocalization of charge among three bipyridine ligands in homoleptic [Ru(bpy)3]2+ during the ligand-centred photoredox processes.22 We speculated that the different electronic structure changes upon photo-excitation of the [Ir(ppy)2(bpy)]+ moiety of SPhos-PC2 and [Ru(bpy)3]2+ moiety of SPhos-PC3 could influence the spatial disposition of α-amino radical formation. Indeed, further investigation led to the discovery of SPhos-PC3 as a highly reactive and regioselective catalyst. The allenylation product 3 was produced predominantly in 71% yield. In comparison, incorporation of a direct covalent bond linking [Ru(bpy)3]2+ and SPhos in SPhos-PC3 overrides the innate regioselectivity of the corresponding dual catalyst system (Scheme 2b). We observed homocoupling of 2 and concurrent oxidation of 1 as the main side reactions.
With the optimal catalyst system in hand, we expanded the substrate scope of the C–H allenylation reaction (Scheme 4). First, we investigated the effects of varying the terminal substituent of propargylic acetate derivatives. Substrates incorporating primary, secondary, and tertiary alkyl substituents such as methyl, cyclohexyl, tert-butyl and 4-chlorobutyl groups all participated in the transformation (5–8, 41–65% yield). Clearly, the steric effect does not play the dominant role in the regioselectivity control. The reaction using a propargylic acetate derivative bearing a terminal allyl group produced the allene product in 39% yield with the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C double bond intact (9). In addition, when the cyclopropyl group was incorporated (10), the desired allene product was obtained in 58% yield. We did not detect the formation of any side product resulting from the opening of a three-membered ring via the propargyl radical.15 Furthermore, the transformation tolerates the terminal phenyl substituents, affording the allenylation product in 60% yield (11).
C double bond intact (9). In addition, when the cyclopropyl group was incorporated (10), the desired allene product was obtained in 58% yield. We did not detect the formation of any side product resulting from the opening of a three-membered ring via the propargyl radical.15 Furthermore, the transformation tolerates the terminal phenyl substituents, affording the allenylation product in 60% yield (11).
 | ||
| Scheme 4 The substrate scope of the C–H allenylation reactions. Reaction conditions: N-aryl tetrahydroisoquinolines (0.3 mmol), propargylic acetate derivatives (2.0 equiv), Pd(OAc)2 (2.0 mol%), SPhos-PC3 (1.0 mol%), 2,6-lutidine (2.0 equiv), CH3CN (0.1 M), 19 W blue LEDs. Isolated yields reported. The diastereomers (1:1–1:1.4 dr) were separated using reverse phase preparative HPLC for characterization (see the ESI† for details). | ||
Moreover, the transformation is highly adaptable to propargylic acetate derivatives bearing various aryl groups. Both electron-donating and electron-withdrawing substituted phenyl groups are well tolerated (12–17). The transformation also provides access to allene products containing a naphthyl group (18) and a range of heteroaromatic rings including benzothiophene (19), furan (20), and thiophene (21). However, the reaction did not convert the substrate bearing a pyridyl group, possibly due to the deleterious effect by the coordination of pyridine to the Pd centre.
We subsequently surveyed a broad spectrum of N-aryl tetrahydroisoquinolines. Substrates incorporating various groups on the N-aryl moiety, irrespective of their electronic properties and substituent patterns, all participate in the C–H allenylation reaction, delivering allene products in 54–73% yield (22–28). To our delight, a C(sp2)–Cl bond was well tolerated (24), although the C(sp2)–Br analogue failed to undergo the reaction. The competitive oxidative addition of the C(sp2)–Br bond likely inhibits the reaction between the Pd centre and propargylic acetate. Additionally, the C–H allenylation reactions of substrates bearing an N-naphthyl group (29) and methoxy-substituted tetrahydroisoquinoline (30) furnished the desired allene products in 60% and 55% yield, respectively.
Product formation was not observed in the absence of Pd(OAc)2 or without irradiation of blue LEDs, indicating that both Pd catalysis and photoredox catalysis are essential. Based on previous work,15,17,23 we postulated a tentative catalytic cycle to illustrate the role of the tethered catalysts (Scheme 5). The reaction is initiated by adduct formation between the Pd centre and 2 (i). Upon photoexcitation of the Ru centre (ii), an SET process with 1 is induced (iii). Hypothetically, a subsequent intramolecular SET process gives rise to a Pd(I) complex (iv), which undergoes bond formation by engaging the α-amino radical intermediate (v).
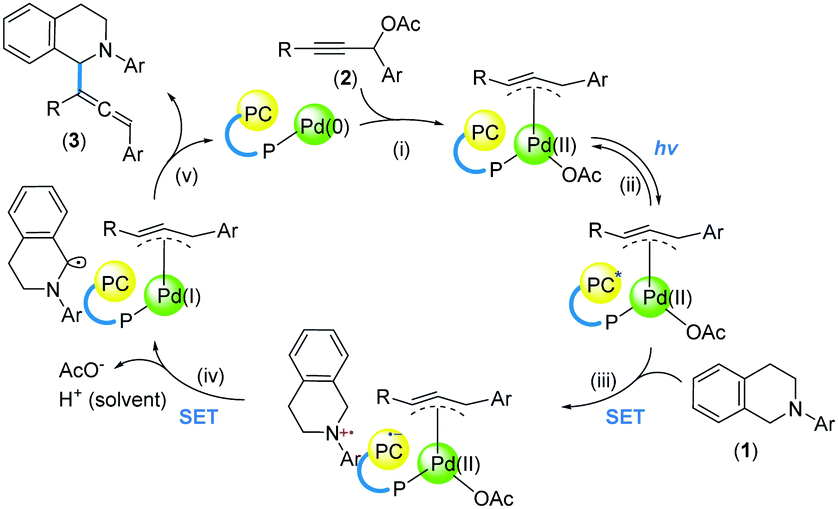 | ||
| Scheme 5 A proposed catalytic cycle of C–H allenylation reactions. | ||
Control experiments showed that the C–O coupling product was not observed when TEMPO was added into the reaction mixture. Coupled with the absence of the ring-opening product of the cyclopropyl substituent (Scheme 4, 10), a propargyl radical, as proposed under dual catalysis (Scheme 1B, b),15 might not be present using the tethered catalysts. This is possibly attributable to the formation of Pd(I) species through an intramolecular SET process24 within the tethered catalysts (Scheme 5, iv).
Besides, we strived to gain structural information to probe the bifunctionality of the tethered catalysts. The conformation revealed by X-ray crystallography (Scheme 3B) suggests that upon coordination to Pd, the Pd centre is likely positioned in proximity to the photocatalyst, and in a sterically unsymmetrical environment. Such spatial arrangements defined by a rigid ligand scaffold plausibly elicit regioselectivity by inducing proximity and orientation effects (Scheme 5, v).
In summary, we have developed a Pd-catalysed C–H allenylation of N-aryl tetrahydroisoquinolines by exploiting the engineered bifunctionality of Pd–SPhos–[Ru(bpy)3] catalysts. We have realised a new regioselectivity control strategy through innovating covalently tethered transition metal–photocatalysts. Given the immense diversity in photocatalysts and transition metal catalysts, we anticipate that pursuing this class of bifunctional catalysts and gaining mechanistic understanding would provide chemists with novel tools to harness transient radical intermediates in transition metal catalysis.
Support from the National University of Singapore (NUS start-up research grant/R-143-000-A52-133) and the Singapore Ministry of Education (Academic research fund/R-143-000-B61-114 and R-143-000-B45-112) is gratefully acknowledged. M. L. thanks NUS for the research scholarship. X. L. C. thanks the SGUnited Traineeships Programme for the additional support. We thank Geok Kheng Tan for solving the X-ray crystal structures.
Conflicts of interest
There are no conflicts to declare.References
- R. Banerjee, Chem. Rev., 2003, 103, 2081 CrossRef CAS.
- N. Shibata and T. Toraya, J. Biochem., 2015, 158, 271 CrossRef CAS PubMed.
- A. Bauer, F. Westkamper, S. Grimme and T. Bach, Nature, 2005, 436, 1139 CrossRef CAS PubMed.
- (a) J. J. Murphy, D. Bastida, S. Paria, M. Fagnoni and P. Melchiorre, Nature, 2016, 532, 218 CrossRef CAS PubMed; (b) T. Zhang, W. Liang, Y. Huang, X. Li, Y. Liu, B. Yang, C. He, X. Zhou and J. Zhang, Chem. Commun., 2017, 53, 12536 RSC; (c) A. Gualandi, F. Calogero, A. Martinelli, A. Quintavalla, M. Marchini, P. Ceroni, M. Lombardo and P. G. Cozzi, Dalton Trans., 2020, 49, 14497 RSC.
- J. Lyu, A. Claraz, M. R. Vitale, C. Allain and G. Masson, J. Org. Chem., 2020, 85, 12843 CrossRef CAS PubMed.
- (a) H. Huo, X. Shen, C. Wang, L. Zhang, P. Rose, L. A. Chen, K. Harms, M. Marsch, G. Hilt and E. Meggers, Nature, 2014, 515, 100 CrossRef CAS PubMed; (b) S. X. Lin, G. J. Sun and Q. Kang, Chem. Commun., 2017, 53, 7665 RSC.
- (a) X. Shen, Y. Li, Z. Wen, S. Cao, X. Hou and L. Gong, Chem. Sci., 2018, 9, 4562 RSC; (b) K. Zhang, L. Q. Lu, Y. Jia, Y. Wang, F. D. Lu, F. Pan and W. J. Xiao, Angew. Chem., Int. Ed., 2019, 58, 13375 CrossRef CAS PubMed; (c) K. Zhou, Y. Yu, Y.-M. Lin, Y. Li and L. Gong, Green Chem., 2020, 22, 4597 RSC.
- (a) Y. Tan, J. M. Muñoz-Molina, G. C. Fu and J. C. Peters, Chem. Sci., 2014, 5, 2831 RSC; (b) H. D. Xia, Z. L. Li, Q. S. Gu, X. Y. Dong, J. H. Fang, X. Y. Du, L. L. Wang and X. Y. Liu, Angew. Chem., Int. Ed., 2020, 59, 16926 CrossRef CAS PubMed; (c) Y. Mao, W. Zhao, S. Lu, L. Yu, Y. Wang, Y. Liang, S. Ni and Y. Pan, Chem. Sci., 2020, 11, 4939 RSC.
- (a) X. Y. Yu, Q. Q. Zhao, J. Chen, J. R. Chen and W. J. Xiao, Angew. Chem., Int. Ed., 2018, 57, 15505 CrossRef CAS PubMed; (b) Q. Guo, M. Wang, Q. Peng, Y. Huo, Q. Liu, R. Wang and Z. Xu, ACS Catal., 2019, 9, 4470 CrossRef CAS; (c) P. Bellotti, M. Koy, C. Gutheil, S. Heuvel and F. Glorius, Chem. Sci., 2021, 12, 1810 RSC.
- Z. Cao, J. Li, Y. Sun, H. Zhang, X. Mo, X. Cao and G. Zhang, Chem. Sci., 2021, 12, 4836 RSC.
- (a) K. Mori, M. Kawashima and H. Yamashita, Chem. Commun., 2014, 50, 14501 RSC; (b) K. Murata, K. Saito, S. Kikuchi, M. Akita and A. Inagaki, Chem. Commun., 2015, 51, 5717 RSC; (c) S.-Y. Yao, M.-L. Cao and X.-L. Zhang, RSC Adv., 2020, 10, 42874 RSC; (d) D. Wang, R. Malmberg, I. Pernik, S. K. K. Prasad, M. Roemer, K. Venkatesan, T. W. Schmidt, S. T. Keaveney and B. A. Messerle, Chem. Sci., 2020, 11, 6256 RSC.
- (a) W.-M. Cheng and R. Shang, ACS Catal., 2020, 10, 9170 CrossRef CAS; (b) C. Zhu, H. Yue, L. Chu and M. Rueping, Chem. Sci., 2020, 11, 4051 RSC; (c) M. Shee and N. D. P. Singh, Catal. Sci. Technol., 2021, 11, 742 RSC; (d) A. Y. Chan, I. B. Perry, N. B. Bissonnette, B. F. Buksh, G. A. Edwards, L. I. Frye, O. L. Garry, M. N. Lavagnino, B. X. Li, Y. Liang, E. Mao, A. Millet, J. V. Oakley, N. L. Reed, H. A. Sakai, C. P. Seath and D. W. C. MacMillan, Chem. Rev., 2022, 122, 1485 CrossRef CAS PubMed.
- (a) H.-M. Huang, P. Bellotti, J. E. Erchinger, T. O. Paulisch and F. Glorius, J. Am. Chem. Soc., 2022, 144, 1899 CrossRef CAS PubMed; (b) H.-M. Huang, P. Bellotti, P.-P. Chen, K. N. Houk and F. Glorius, Nat. Synth., 2022, 1, 59 CrossRef; (c) H.-M. Huang, P. Bellotti, C. G. Daniliuc and F. Glorius, Angew. Chem., Int. Ed., 2021, 60, 2464 CrossRef CAS PubMed.
- B. Niu, Y. Wei and M. Shi, Org. Chem. Front., 2021, 8, 3475 RSC.
- Z. Z. Zhou, R. Q. Jiao, K. Yang, X. M. Chen and Y. M. Liang, Chem. Commun., 2020, 56, 12957 RSC.
- (a) Z. Z. Zhou, X. R. Song, S. Du, K. J. Xia, W. F. Tian, Q. Xiao and Y. M. Liang, Chem. Commun., 2021, 57, 9390 RSC; (b) Y. Chen, K. Zhu, Q. Huang and Y. Lu, Chem. Sci., 2021, 12, 13564 RSC; (c) Y. Chen, J. Wang and Y. Lu, Chem. Sci., 2021, 12, 11316 RSC.
- (a) J. Xuan, T. T. Zeng, Z. J. Feng, Q. H. Deng, J. R. Chen, L. Q. Lu, W. J. Xiao and H. Alper, Angew. Chem., Int. Ed., 2015, 54, 1625 CrossRef CAS PubMed; (b) K. C. Cartwright and J. A. Tunge, Chem. Sci., 2020, 11, 8167 RSC; (c) H.-M. Huang, P. Bellotti and F. Glorius, Chem. Soc. Rev., 2020, 49, 6186 RSC.
- K. Nakajima, Y. Miyake and Y. Nishibayashi, Acc. Chem. Res., 2016, 49, 1946 CrossRef CAS PubMed.
- Y. Zheng, B. Miao, A. Qin, J. Xiao, Q. Liu, G. Li, L. Zhang, F. Zhang, Y. Guo and S. Ma, Chin. J. Chem., 2019, 37, 1003 CrossRef CAS.
- Y. Lou, J. Wei, M. Li and Y. Zhu, J. Am. Chem. Soc., 2022, 144, 123 CrossRef PubMed.
- R. Bevernaegie, L. Marcelis, B. Laramee-Milette, J. De Winter, K. Robeyns, P. Gerbaux, G. S. Hanan and B. Elias, Inorg. Chem., 2018, 57, 1356 CrossRef CAS PubMed.
- E. Medina and B. Pinter, J. Phys. Chem. A, 2020, 124, 4223 CrossRef CAS PubMed.
- (a) H.-M. Huang, P. Bellotti, P. M. Pflüger, J. L. Schwarz, B. Heidrich and F. Glorius, J. Am. Chem. Soc., 2020, 142, 10173 CrossRef CAS PubMed; (b) H.-M. Huang, M. Koy, E. Serrano, P. M. Pflüger, J. L. Schwarz and F. Glorius, Nat. Catal., 2020, 3, 393 CrossRef CAS; (c) K. P. S. Cheung, D. Kurandina, T. Yata and V. Gevorgyan, J. Am. Chem. Soc., 2020, 142, 9932 CrossRef PubMed.
- W. Iali, P.-H. Lanoe, S. Torelli, D. Jouvenot, F. Loiseau, C. Lebrun, O. Hamelin and S. Ménage, Angew. Chem., Int. Ed., 2015, 54, 8415 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2114578 and 2114579. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d2cc01064j |
| This journal is © The Royal Society of Chemistry 2022 |