
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMetallation of sensitive fluoroarenes using a potassium TMP-zincate supported by a silyl(bis)amido ligand†
Pasquale
Mastropierro
a,
Alan R.
Kennedy
 b and
Eva
Hevia
*a
b and
Eva
Hevia
*a
aDepartement für Chemie, Biochemie und Pharmazie, Universität Bern, Switzerland. E-mail: eva.hevia@dcb.unibe.ch
bDepartment of Pure and Applied Chemistry, University of Strathclyde, Glasgow, UK
First published on 9th March 2022
Abstract
Combining a bulky bis(amide) and a reactive one-coordinate TMP (2,2,6,6-tetramethylpiperidide) ligand, a new mixed K/Zn heteroleptic base has been developed for regioselective zincation of fluoroarenes. This special ligand set allows for trapping and structural authentication of the first intermediates of direct Zn–H exchange of fluoroarenes obtained via deprotonative metallation, providing mechanistic insights of the processes involved.
Fluoroarenes represent one of the most ubiquitous fragments present in many biologically active and pharmaceutical compounds.1 Thus developing methods that allow the incorporation of these units into more complex molecular scaffolds is of increasing importance.2 Despite their synthetic relevance, metallation of fluoroarenes using conventional organolithium bases represents a significant challenge due to the high sensitivity of the generated metallo-intermediates and their tendency to undergo decomposition (e.g., benzyne formation, autometallation), even under cryogenic conditions.3 Alternative approaches using bimetallic bases which combine an alkali-metal with a less electropositive metal such as Zn, Al, Ga or Fe have been developed to address some of these limitations.4–6 Within this set, alkali-metal zincates are particularly appealing as they can grant access to valuable fluoroarylzinc intermediates which can then be functionalised further via C–C bond forming processes.7 For example, Knochel has capitalised on the mild metallating ability of (TMP)ZnCl·LiCl (TMP = 2,2,6,6-tetramethylpiperidide) to perform zincation of sensitive aromatic substrates including fluoroarenes at room temperature.4c Similarly, Kondo and Uchiyama have demonstrated the reactivity of lithium zincates LiZn(TMP)R2 with haloarenes can be finely tuned depending on the nature of R, to produce polyfunctional haloarenes via zincation/electrophilic interception when R = tBu; whereas if R = Me, benzyne intermediates are formed which can be trapped by dienes in Diels–Alder reactions.4a While these studies show the practicality of lithium zincates for metallation of fluoroarenes, no experimental evidence has been collected on the chemistry taking place between the limits of the starting materials and the zinc-free quenched products. An alternative method for zincation of fluoroarenes has recently been developed by Crimmin combining stoichiometric amounts of a zinc hydride complex supported by a sterically demanding kinetically stabilising β-diketiminate ligand with Pd catalysis.8 Insightful mechanistic studies have revealed that these reactions occur via the formation of Pd/Zn heterobimetallic species.
Breaking new ground in this field, here we report a new bimetallic base, which combining the kinetically activated TMP with a bulky silyl(bis)amide ligand, enables the regioselective zincation of fluoroarenes and the trapping and characterization of the relevant zinc-containing intermediates.
Building on our recent studies investigating the synthesis of reactivity of alkali-metal zincates containing the bulky dianionic silyl(bis)amide ligand {Ph2Si(NAr*)2}2− (Ar* = 2,6-diisopropylphenyl),9 the new potassium zincate [{Ph2Si(NAr*)2Zn(TMP)}−{K(THF)6}+] (1) was prepared via stepwise two-fold deprotonation of Ph2Si(NHAr*)2 using single metal bases KCH2SiMe3 and Zn(TMP)2 in a sequential manner (Fig. 1).
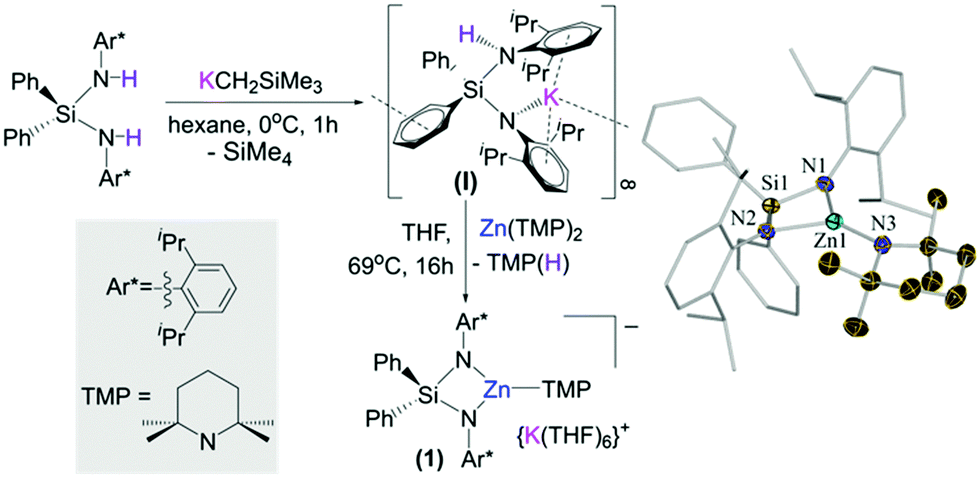 | ||
| Fig. 1 Stepwise synthesis of [{Ph2Si(NAr*)2Zn(TMP)}−{K(THF)6}+] (1). Molecular structure of the anion present in 1. Thermal ellipsoids are rendered at 50% probability. Hydrogen atoms are omitted and Ph and Ar* fragments are drawn as wire frames for clarity. | ||
While the first metallation to give [Ph2Si(NHAr*)(NAr*)K]∞ (I) took place rapidly (0 °C, 2 h),9b the subsequent zincation of the remaining NHAr* group required forcing conditions (69 °C, 16 h). Attempts to prepare 1 by reacting the silyl(bis)amine precursor with an equimolar mixture of KCH2SiMe3 and Zn(TMP)2 led to the isolation of [{Ph2Si(NAr*)2Zn(CH2SiMe3)}−{K(THF)4}+] (2) (see ESI†).
We next assessed the reactivity of 1 towards 1,3,5-trifluorobenzene as a model fluoroarene. 1H NMR monitoring of the reaction in d8-THF solution showed evidence of metallation at room temperature although the reaction was slow, taking three days to reach completion. Reaction times could be reduced significantly by heating to refluxing conditions affording [{Ph2Si(NAr*)2Zn(C6H2F3)}−{K(THF)6}+] (3a) in a 90% yield in three hours (Fig. 2). In contrast, evidencing the kinetic activation of 1, 1,3,5-trifluorobenzene is inert towards zincation using single metal Zn(TMP)2. Notwithstanding, the TMP amide group seems to be key for the success of the reaction since potassium alkylzincate 2 failed to react with this fluoroarene after 24 h at 69 °C. It should also be noted that reaction conditions required to form 3 are significantly milder than those reported for the zincation of 1,3,5-trifluorobenzene using Pd-catalysis (up to 6 days at 50/80 °C).8
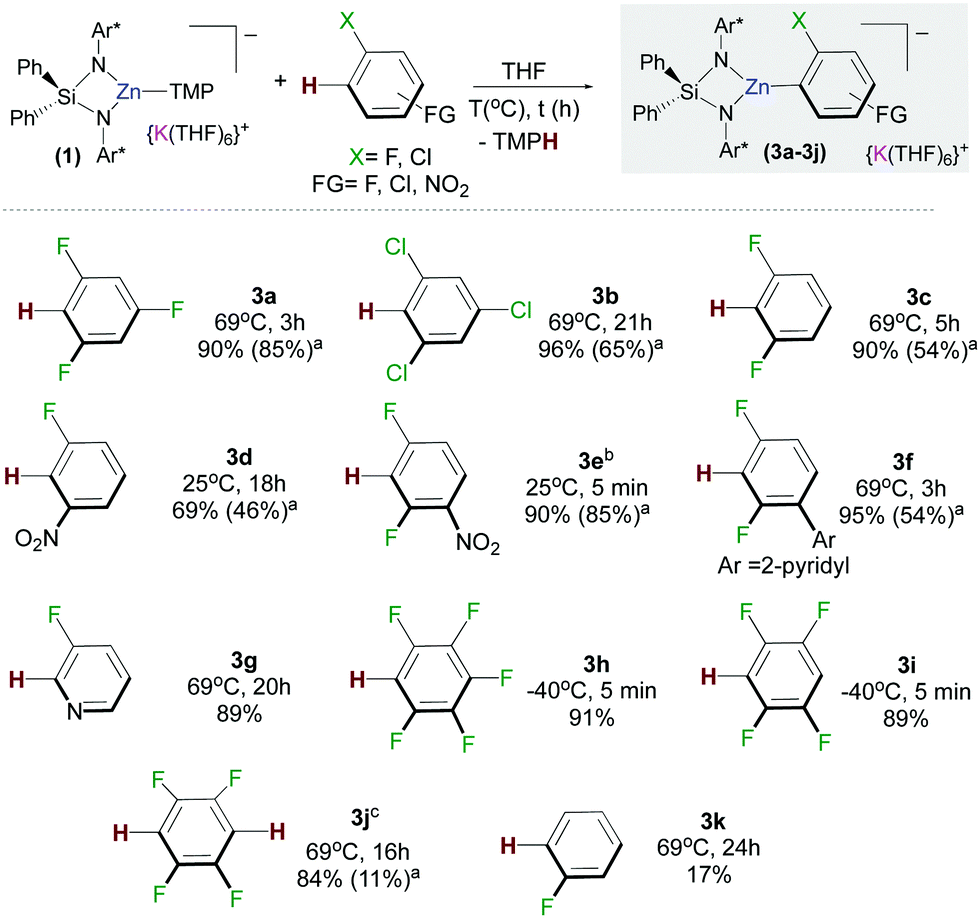 | ||
Fig. 2 Scope of the C–H zincation of fluoroarenes using potassium zincate 1. NMR yields determined by 1H NMR spectroscopic data using hexamethylbenzene as an internal standard. a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) Isolated crystalline yields. bCompound 2e is light sensitive. cReaction carried out using 0.5 equivalents of 1,2,4,5-tetrafluorobenzene. Isolated crystalline yields. bCompound 2e is light sensitive. cReaction carried out using 0.5 equivalents of 1,2,4,5-tetrafluorobenzene. | ||
Trying to ascertain the role of the potassium in the reaction producing 3a, metallation studies were repeated using the sodium zincate congener [{Ph2Si(NAr*)2Zn(TMP)}−{Na(THF)6}+] and using 1 in the presence of macrocyclic Lewis donor [2.2.2]-cryptand. The latter has the ability to coordinatively trap and sequester the K cation (see ESI† for details).10 In each case no significant differences between the yields, reaction times and selectivity were observed from those reported for 3a when using 1. This is somewhat surprising since many studies on alkali-metal ates have unearthed a marked alkali-metal effect in the reactivity of such heterobimetallic complexes.11 Furthermore, mechanistic studies on the ferration of fluoroarenes by sodium trisamidoferrate [NaFe(HMDS)3] (HMDS = N(SiMe3)2) have revealed that initial coordination of the substrate to the alkali-metal via Na⋯F interactions is key for the reaction to take place, which involves a two-step mechanism with initial Na–H exchange followed by fast intramolecular transmetallation to iron.6c Interestingly, here, for the zincation of 1,3,5 trifluorobenzene potassium appears to play a secondary role, stabilizing the zincate anion intermediates. These findings suggest that the enhanced basicity of 1 may mainly be due to the kinetic activation of the zincate anion when compared to more conventional neutral zinc amide bases.
The scope of this approach was then further investigated (Fig. 2). 1,3,5-Trichlorobenzene and 1,3-difluorobenzene can also be zincated by 1 furnishing 3b and 3c in 96 and 90% yields respectively, although longer reaction times are required. Reflecting the stabilizing effect of the bulky {Ph2Si(NAr*)2}2−, the formation of 3b under these conditions (21 h, 69 °C) contrasts with previous studies by Mulvey on the zincation of chlorobenzene by [(TMEDA)NaZn(TMP)tBu2], where the relevant metallated intermediate undergoes rapid NaCl elimination forming a benzyne species that can be trapped by tBu2Zn·TMEDA.12 Encouraged by these findings we next pondered if this approach could also tolerate nitro groups. Previous work by Knochel4c,13 has shown promise on the compatibility of these bimetallic reagents with this highly sensitive functional group. Pleasingly it was found 1 reacts at room temperature with 3-fluoronitrobenzene and 2,4-difluoronitrobenzene forming 3d and 3e in respective 69% and 90% yields. This approach is also compatible with pyridine-substituted substrates as shown for zincation of 2-(2,4-difluorophenyl)pyridine which takes place regioselectively ortho to both fluorine substituents (3f, 95%). C2-Zincation is also observed for 3-fluoropyridine (3g, 89%) contrasting with previous studies using organolithium reagents or the Lochmann–Schlosser super base reagent were C4-metallation is preferred.14 Reactions of 1 with the more activated substrates15 1,2,4,5-tetrafluorobenzene and pentafluorobenzene furnished zincated products 3h and 3i in 91% and 89% yields respectively; whereas for fluorobenzene low conversions were observed after 24 h at 69 °C (3k in Fig. 2). While 3h and 3i are stable at room temperature, low temperatures (−40 °C) were employed for their synthesis in order to control the TMP-reactivity of 1 (vide infra).
Remarkably in the case of 1,2,4,5-tetrafluorobenzene dizincation can be achieved by using 2 equivalents of 1 (3j, 84%, Fig. 2 and Scheme 1). 1H NMR monitoring of this reaction established its stepwise nature. Formation of mono-zincated product 3i occurs almost instantaneously, which in turn, can be twofold metallated by the second equivalent of 1 (Scheme 1). While this second step requires forcing conditions to achieve good conversions (16 h, 69 °C), these findings show the potential of 1 to promote polyzincation reactions which to the best of our knowledge is unprecedented for fluoroarenes.
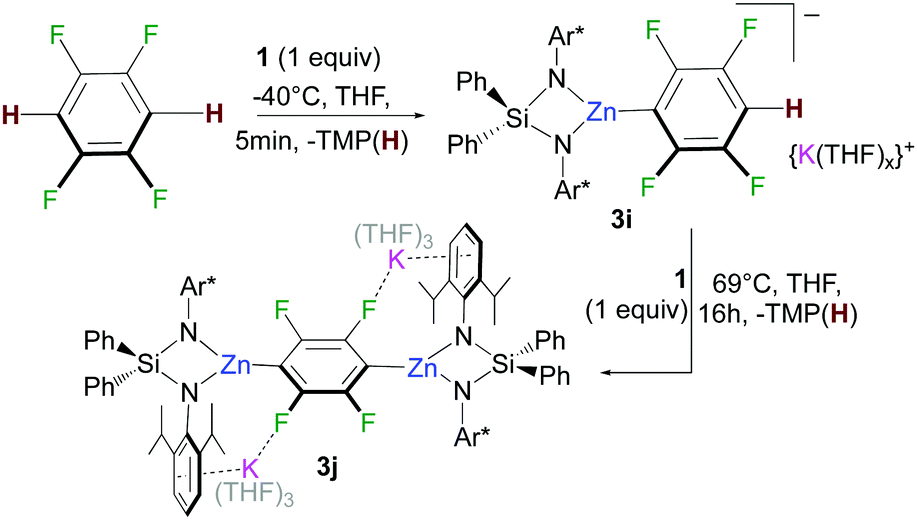 | ||
| Scheme 1 Stepwise 1,4-di-zincation of 1,2,4,5-tetrafluorobenzene by 1. | ||
Compounds 3a–e, 3h and 3j were isolated as crystalline solids and their structures were authenticated by X-ray crystallography (Fig. 3, 4 and ESI†), demonstrating that these compounds are indeed genuine products of Zn–H exchange reactions. Solvent-separated ion pair motifs were found for 3a–d and 3h, comprising, in each case, a potassium cation solvated by six molecules of THF. Their anionic moiety comprises a trigonal planar zinc centre which binds to the bidentate silylbis(amido) ligand and a terminal aryl group. Contrastingly 3e exhibits a contacted ion pair structure, where potassium is now solvated by three THF molecules and interacts with the zincate anion by π-engaging with one of its Ar* rings in a η4-fashion and by forming two further bonds with a F and one nitro O atom (F1 and O1 in Fig. 3c).
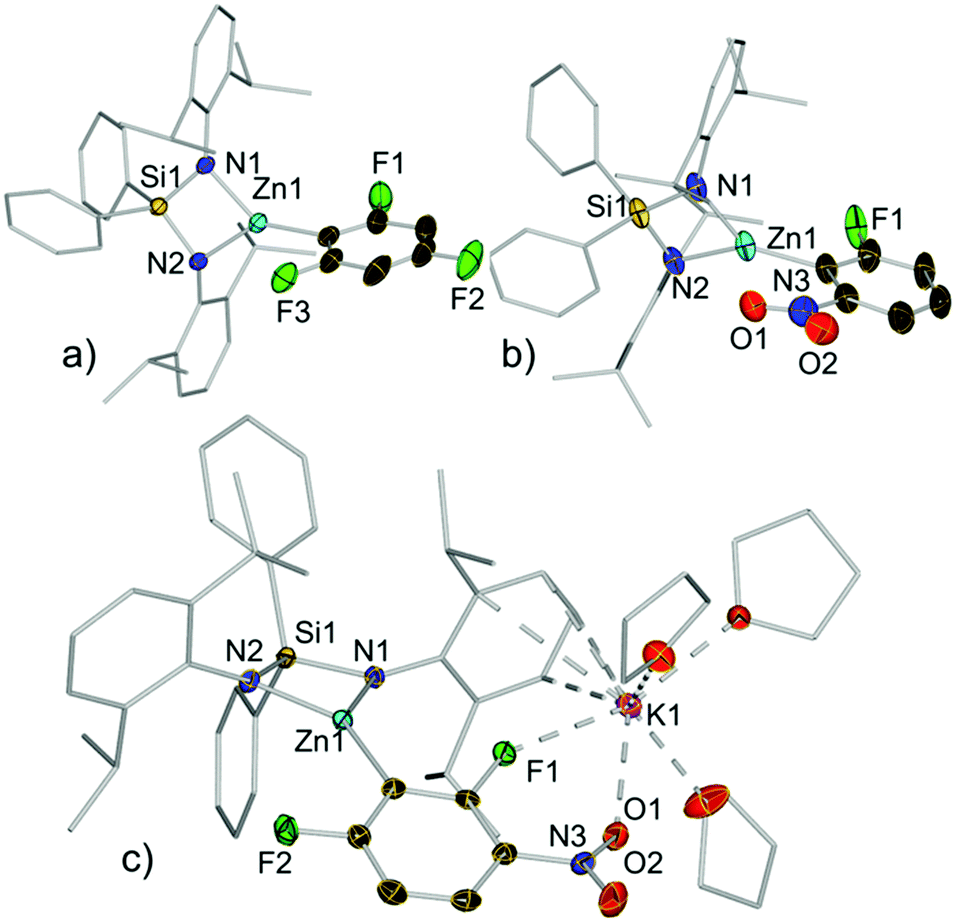 | ||
| Fig. 3 Crystal structures of: (a) anion of 3a; (b) anion of 3d; and (c) 3e. Thermal ellipsoids are rendered at 50% probability. Hydrogen atoms and minor disorder components are omitted and Ph and Ar* fragments and THF molecules are drawn as wire frames for clarity. | ||
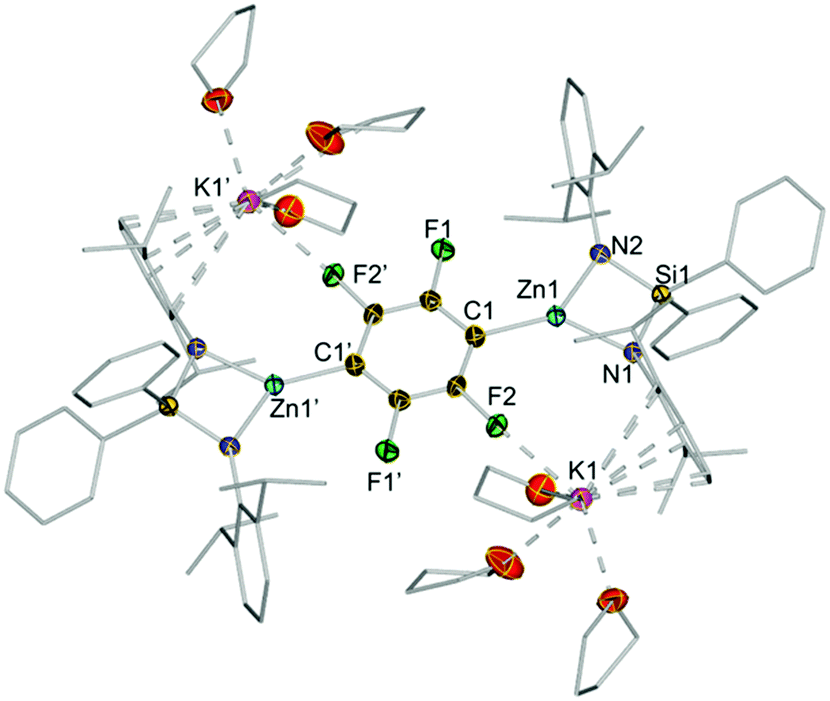 | ||
| Fig. 4 Molecular crystal structures of 3j. Thermal ellipsoids are rendered at 50% probability. Hydrogen atoms and minor disorder components are omitted and Ph and Ar* fragments and THF molecules are drawn as wire frames for clarity. | ||
As far as we can ascertain 3d and 3e constitute the first examples of direct metallation of nitroarenes to be structurally characterized. We attribute their unexpected stability to the chelating bulky bis(amide) ligand which can offer steric protection for the fragile aryl fragments, allowing their isolation and characterization. This was further supported by the fact that attempts to isolate the product of the reaction of 2,4-difluoronitrobenzene with the related potassium zincate [(PMDETA)KZn(TMP)Et2]16 (PMDETA = N,N,N′,N′′,N′′-pentamethylethylenediamine) led to the formation of a complex mixture of decomposition products.
The centrosymmetric molecular structure of 3j confirmed a two-fold deprotonated 1,2,4,5-tetrafluoroaryl ring with two Zn centres occupying the positions previously filled by H atoms (Fig. 4). Solvated by three molecules of THF, each potassium interacts with one F atom of the ring as well as π-engaging with one Ar* group in a η6-fashion, lying essentially in the same plane defined by the fluoroaryl ring. As mentioned before for 3d–e, the chelating silyl(bis)amide ligand may also play an important role by facilitating the formation of this structure providing a protective steric shelter for these newly formed Zn–C bonds.
Despite the unexpected stability of zincated products 3a–j, preliminary reactivity studies have shown that their Zn–C bonds are still reactive enough to participate in C–C bond forming reactions, demonstrating their potential for onward reactivity applications. Thus, 3a undergoes Pd catalysed cross-coupling with 4-bromo-benzonitrile, affording the anticipated bis(aryl) product in a 63% yield. 3a also reacts with benzoyl chloride furnishing PhC(O)ArF (ArF = C6H2F3) in a 70% yield (see ESI†).
In addition, 1H/19F NMR reaction monitoring studies of the reaction of 1 with pentafluorobenzene revealed that at room temperature along with the formation of 3h and TMP(H), variable amounts of Ph2Si(NHAr*)2 and a new organometallic species which also contained {C6F5}− groups could be detected in solution. A similar observation was seen for 1,2,4,5-tetrafluorobenzene. These findings are consistent with potassium zincate 1 exhibiting poly-basicity using its silyl(bis)amide ligand. This was confirmed by repeating the reaction of 1 with a three molar excess of C6F5H which led to the formation and isolation of potassium zincate [Ph2Si(NHAr*)(NAr*)KZn(C6F5)2]∞ (4) as a crystalline solid (Fig. 5). Exhibiting a contacted-ion pair structure, 4 is the result of the metallation of two equivalents of C6F5H by 1 using its TMP base and one amide base unit of its chelating {Ph2Si(NAr*)2}2− ligand. These findings show that while the latter acts as a spectator in most of these reactions (Fig. 3), it can also be basic enough to promote the metallation of particularly activated substrates with strong electron withdrawing substituents such as C6F5H or C6F4H2. The newly generated silyl(amide)amine ligand coordinates to Zn via its amido N in a monodentate fashion {Ph2Si(NHAr*)(NAr*)} whereas K π-bonds with four aromatic carbons of the Ar* ring located on the amido N (Fig. 5). Zn displays a distorted trigonal planar geometry binding to two C6F5 aryls, forming two strong sigma bonds with the metallated carbons. K completes its coordination by binding to one THF molecule and forming five additional K⋯F contacts. One F belongs to a C6F5 ring attached to Zn1 (F6 in Fig. 5) whereas the other four belong to two different C6F5 fragments from a neighbouring unit (F4′′ and F5′′; F1′′′ and F2′′′ in Fig. 5), giving rise to an intricate 3D polymeric structure (see ESI†).17 Compound 4 co-crystallizes with variable amounts of Ph2Si(NHAr*)2 resulting from the remaining amido group in 3 acting as a base towards C6F5H. Consistent with this interpretation the 19F NMR spectra of the reaction crudes show an additional species that we tentatively assign as the tris(fluoroaryl) zincate [(THF)xKZn(C6F5)3] (see ESI† for details).
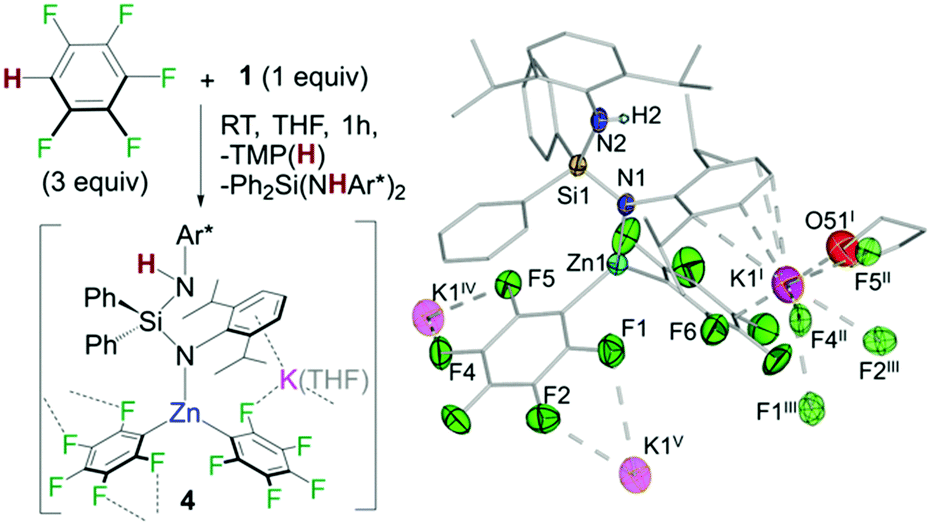 | ||
| Fig. 5 Reaction of 1 with a three-molar excess of pentafluorobenzene. Molecular structure of 4 thermal ellipsoids are rendered at 50% probability. Hydrogen atoms are omitted and Ph and Ar* fragments and THF molecules are drawn as wire frames for clarity. | ||
To conclude, a new mixed K/Zn base has been developed for the regioselective zincation of a range of fluoroarenes, including challenging substrates containing sensitive NO2 groups. Reactions give good yields with excellent control of regioselectivity. Key for the success of this approach seems to be the combination of a sterically demanding bis(amide) supporting ligand, which provides stability to the fragile fluoroaryl fragments in the metallated intermediates; and a terminal, one-coordinate, kinetically activated TMP basic site on 1, which enables direct zincation of the substrates. Combining X-ray crystallographic studies with NMR monitoring of the reactions, the first structural and mechanistic insights into these Zn–H exchanges processes have been gained. Preliminary reactivity studies also show that these organometallic compounds can engage in onward C–C bond forming processes.
We thank the SNSF (188573) and the University of Bern for their generous sponsorship of this research.
Conflicts of interest
There are no conflicts to declare.Notes and references
- (a) J. Wang, M. Sánchez-Roselló, J. Aceña, C. Del Pozo, A. E. Sorochinsky, S. Fustero, V. A. Soloshonok and H. Liu, Chem. Rev., 2014, 114, 2432 CrossRef CAS PubMed; (b) S. Purser, P. R. Moore, S. Swallow and V. Gouverneur, Chem. Soc. Rev., 2008, 37, 320 RSC.
- T. Ahrens, J. Kohlmann, M. Ahrens and T. Braun, Chem. Rev., 2015, 115, 931 CrossRef CAS PubMed.
- (a) L. Gupta, A. C. Hoepker, K. J. Singh and D. B. Collum, J. Org. Chem., 2009, 74, 2231 CrossRef CAS PubMed; (b) M. Schlosser, L. Guio and F. Leroux, J. Am. Chem. Soc., 2001, 123, 3822 CrossRef CAS PubMed; (c) J. Clayden, Organolithiums: Selectivity for Synthesis, Pergamon, Oxford, 2002 Search PubMed.
- (a) M. Uchiyama, T. Miyoshi, Y. Kajihara, T. Sakamoto, Y. Otani, T. Ohwada and Y. Kondo, J. Am. Chem. Soc., 2002, 124, 8514 CrossRef CAS PubMed; (b) S. Wunderlich and P. Knochel, Angew. Chem., Int. Ed., 2007, 46, 7685 CrossRef CAS PubMed; (c) M. Mosrin and P. Knochel, Org. Lett., 2009, 11, 1837 CrossRef CAS PubMed.
- (a) S. Wunderlich and P. Knochel, Angew. Chem., Int. Ed., 2009, 48, 1501 CrossRef CAS PubMed; (b) R. McLellan, M. Uzelac, A. R. Kennedy, E. Hevia and R. E. Mulvey, Angew. Chem., Int. Ed., 2017, 56, 9566 CrossRef CAS PubMed.
- (a) S. H. Wunderlich and P. Knochel, Angew. Chem., Int. Ed., 2009, 48, 9717 CrossRef CAS PubMed; (b) L. C. H. Maddock, T. Nixon, A. R. Kennedy, M. R. Probert, W. Clegg and E. Hevia, Angew. Chem., Int. Ed., 2018, 57, 187 CrossRef CAS PubMed; (c) L. C. H. Maddock, M. Mu, A. R. Kennedy, M. García-Melchor and E. Hevia, Angew. Chem., Int. Ed., 2021, 60, 15296–15301 CrossRef CAS PubMed.
- Y. Okamoto, K. Yoshioka, T. Yamana and H. Mori, J. Organomet. Chem., 1989, 369, 285 CrossRef CAS.
- M. Garçon, N. W. Mun, A. J. P. White and M. R. Crimmin, Angew. Chem., Int. Ed., 2021, 60, 614 CrossRef PubMed.
- (a) P. Mastropierro, Z. Livingstone, S. D. Robertson, A. R. Kennedy and E. Hevia, Organometallics, 2020, 39, 4273 CrossRef CAS; (b) P. Mastropierro, A. R. Kennedy and E. Hevia, Eur. J. Inorg. Chem., 2021, 1016 CrossRef CAS.
- J. Hicks, P. Vasko, J. M. Goicoechea and S. Aldridge, J. Am. Chem. Soc., 2019, 141, 11000 CrossRef CAS PubMed.
- (a) T. X. Gentner and R. E. Mulvey, Angew. Chem., Int. Ed., 2021, 60, 9247 CrossRef CAS PubMed; (b) T. X. Gentner, M. J. Evans, A. R. Kennedy, S. E. Neale, C. L. McMullin, M. P. Coles and R. E. Mulvey, Chem. Commun., 2022, 58, 1390 RSC.
- D. R. Armstrong, L. Balloch, W. Clegg, S. H. Dale, P. Garcia-Alvarez, E. Hevia, L. M. Hogg, A. R. Kennedy, R. E. Mulvey and C. T. O’Hara, Angew. Chem., Int. Ed., 2009, 48, 8675 CrossRef CAS PubMed.
- M. Balkenhohl, D. S. Ziegler, A. Desaintjean, L. J. Bole, A. R. Kennedy, E. Hevia and P. Knochel, Angew. Chem., Int. Ed., 2019, 58, 12898 CrossRef CAS PubMed.
- (a) F. Marsais and G. Queginer, Tetrahedron, 1983, 39, 2009 CrossRef CAS; (b) G. Shi, S. Takagishi and M. Schlosser, Tetrahedron, 1994, 50, 1129 CrossRef CAS.
- H. H. Büker, N. M. M. Nibbering, D. Espinosa, F. Mongin and M. Schlosser, Tetrahedron Lett., 1997, 38, 8519 CrossRef.
- B. Conway, D. V. Graham, E. Hevia, A. R. Kennedy, J. Klett and R. E. Mulvey, Chem. Commun., 2008, 2638 RSC.
- For a review on the coordination chemistry of the CF unit in fluorocarbons, see: H. Plenio, Chem. Rev., 1997, 97, 3363 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2151990–2152000. For ESI and crystallographic data in CIF or other electronic format see https://doi.org/10.1039/d2cc00979j |
| This journal is © The Royal Society of Chemistry 2022 |