
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTotal syntheses of strained polycyclic terpenes†
Gleb A.
Chesnokov
 and
Karl
Gademann
*
and
Karl
Gademann
*
Department of Chemistry, University of Zurich, Winterthurerstrasse 190, CH-8057 Zurich, Switzerland. E-mail: karl.gademann@uzh.ch
First published on 30th March 2022
Abstract
Terpenoids constitute a broad class of natural compounds with tremendous variability in structure and bioactivity, which resulted in a strong interest of the chemical community to this class of natural products over the last 150 years. The presence of strained small rings renders the terpenoid targets interesting for chemical synthesis, due to limited number of available methods and stability issues. In this feature article, a number of recent examples of total syntheses of terpenoids with complex carbon frameworks featuring small rings are discussed. Specific emphasis is given to the new developments in strategical and tactical approaches to construction of such systems.
 Gleb A. Chesnokov | Gleb A. Chesnokov (1995) received his Diploma in Chemistry in 2019 from Lomonosov Moscow State University, Moscow, Russia. Then he joined the group of Prof. Karl Gademann, and currently is working in the field of total synthesis of complex natural products. |
 Karl Gademann | Karl Gademann (1972) was educated at ETH Zürich (D. Seebach, E. M. Carreira) and Harvard University (E. N. Jacobsen). Previous professorial appointments include the EPFL in Lausanne and the University of Basel, and since 2016, as a full professor at the University of Zurich. Karl Gademann is a member of the research council of the Swiss National Science Foundation. His work has been recognized by a number of international awards: the European Young Investigator Award, the Latsis prize, the Novartis Early Career Award, the Ružička Medal, the Liebig Lecture Award, and he was elected as a Fellow of Chemistry Europe. |
Introduction
Terpenes have fuelled discovery in society and science for decades. Terpene-derived vitamins, fragrances, drugs, pigments, and materials cured, cared, and coloured our society and our lives. Progress in science was driven by the complex molecular architecture of terpenes, their potent bioactivity, and their economic potential over the last century up to this decade.Synthetic chemistry greatly was inspired and challenged by the often intricate and elaborate carbon framework of terpenes and total synthesis as a research approach, the development of synthetic and analytical methods, correlation through chemical degradation, and tactics and strategies were constantly improved.
Over the last decade, many novel tactics and strategies in terpene synthesis were developed and powerful new methods were put to the acid test on complex terpenoid structures; and the progress was reviewed in many excellent articles.1 It is the goal of this feature article to discuss and to highlight recent examples in the total synthesis of polycyclic terpenoids featuring small rings. These cyclopropane and cyclobutane derivatives often pose rather different constraints on both methods and on tactics and strategies, due to ring strain, the associated higher energy, and undesired side-reactions such as cyclopropane opening or rearrangements.
Additionally, we provide problem-sets associated with these syntheses in the ESI.† These questions could be used by interested readers or in group meetings to learn and to discuss chemical challenges and their solutions. We hope the readers will find these useful and that these examples also benefit the educational aspects of this feature article. It is a part of our larger efforts to contributing to the education of the community by providing materials on complex molecular assembly problems.
Terpenoids with small rings
Overview of different targets
Total synthesis of terpenoids is a highly dynamic field with dozens of targets both classical and newly isolated being synthesised every year. In order to put the small ring containing terpenoids in a broader context, Chart 1 summarises a number of polycyclic terpenes2–36 prepared over the last 5 years, highlighting key reactions, utilised to form the corresponding ring systems. This chart is intended to give a reader an overview of modern approaches to construction of polycyclic carbon architectures in modern synthetic chemistry. While it is beyond the scope of this article to discuss these synthetic endeavours, the reader is encouraged to read the original literature, if a natural product and its associated complex structure catches the interest. In the following sections, a portfolio of strained polycyclic terpenes syntheses will be discussed as case studies. | ||
| Chart 1 Selection of polycyclic terpenoids synthesised over the last 5 years with carbon core and key cycle closing reactions highlighted. | ||
ent-Shagenes
Shagenes A (2) and B (46) are marine tricyclic terpenoids, discovered in 2014 by Baker and co-workers,37 featuring a unique 3/6/5-membered ring system. These interesting targets were synthesised by Tsukano, Takemoto, and co-workers in 2021 (Scheme 1).3 One particular challenge in this polycyclic terpenoid involving the cyclopropane unit was the presence of a vinyl substituent, which potentially could lead to instability during chemical transformations. Three all-carbon rings were forged in only two chemical steps, showcasing fast and controlled construction of molecular complexity.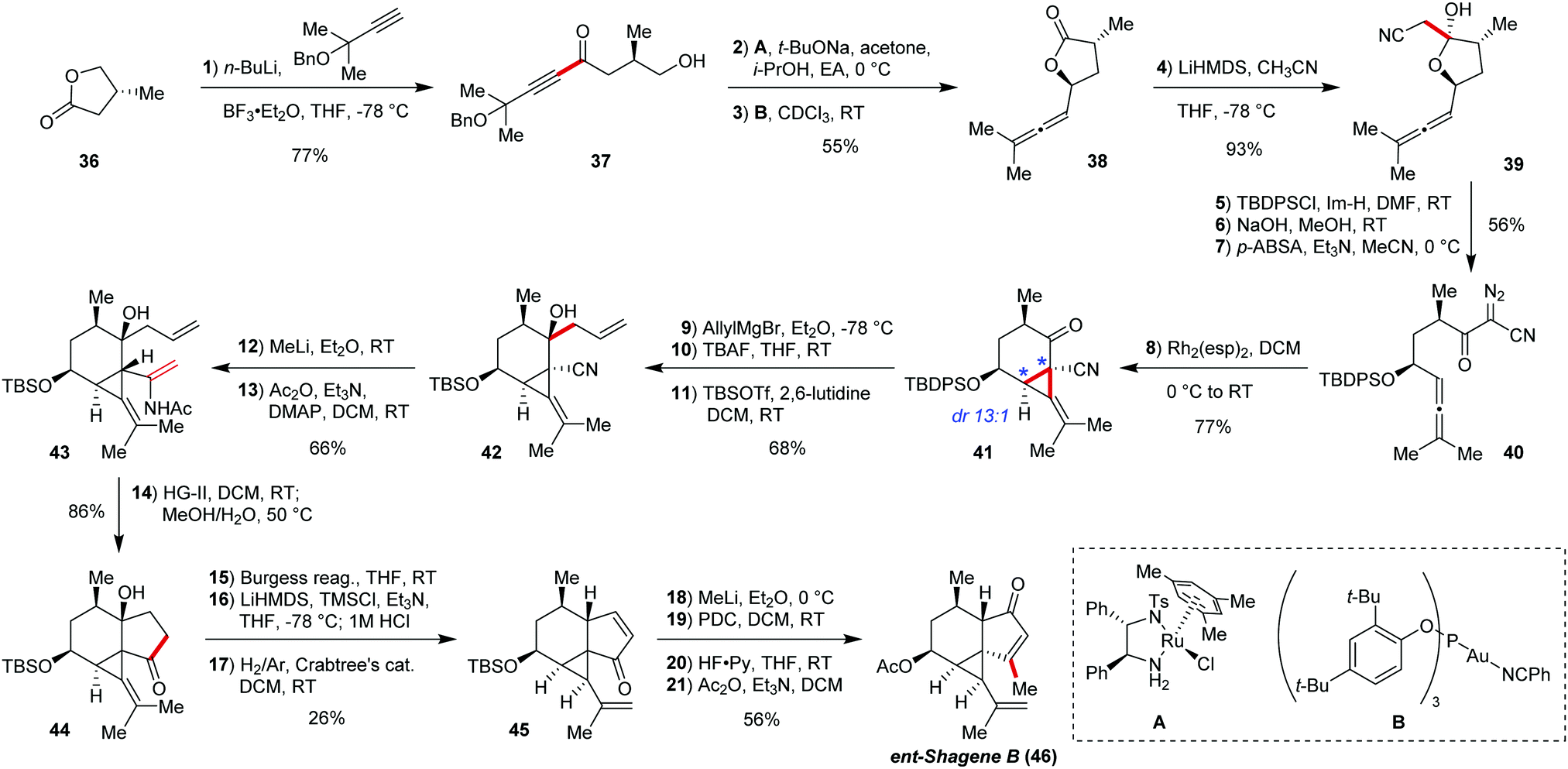 | ||
| Scheme 1 Total synthesis of ent-shagenes A and B by Tsukano, Takemoto and co-workers. | ||
The synthesis starts with enantiopure lactone 36 (prepared via Helmchen's approach38), which was opened by addition of lithiated benzyl ether of 2-ethyl-3-butyn-2-ol. The obtained ketoalcohol 37 was selectively closed back to lactone by hydroacylation with Noyori's transfer hydrogenation catalyst (dr 18![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1).39 The propargyl ether side chain was converted to the corresponding allene by a Au-catalyzed 1,5-hydride shift/fragmentation sequence40 to give 38. Subsequent addition of the acetonitrile conjugate base to the lactone 38, followed by hydroxy group protection with TBDPSCl, cleavage of the intermediate Si enol ether with base, and Regitz diazo transfer by 4-acetamidobenzenesulfonyl azide (p-ABSA) furnished formation of the compound 40, the substrate for the key cyclisation step of the synthesis.
:1).39 The propargyl ether side chain was converted to the corresponding allene by a Au-catalyzed 1,5-hydride shift/fragmentation sequence40 to give 38. Subsequent addition of the acetonitrile conjugate base to the lactone 38, followed by hydroxy group protection with TBDPSCl, cleavage of the intermediate Si enol ether with base, and Regitz diazo transfer by 4-acetamidobenzenesulfonyl azide (p-ABSA) furnished formation of the compound 40, the substrate for the key cyclisation step of the synthesis.
The intramolecular cyclopropanation was found to be the most efficient under Rh2(esp)2 catalysis (5 mol%) providing the bicyclic intermediate 41 with the tetrasubstituted cyclopropane in 77% yield and high diastereoselectivity (dr 13:1). The presence of the TBDPS protecting group on the hydroxy group in 40 was crucial for high selectivity, as the selectivity dropped to 3.4:1 with the TBS protected substrate. Successful and stereocontrolled formation of the bicyclic core allowed further modifications in order to set the stage for the ring closing metathesis (RCM) based construction of the 5-membered cycle. The product 41 was treated with allyl magnesium bromide solution, followed by protection group exchange (to TBS). Then, the nitrile group in 42 was converted to an acetenamide by methyllithium addition and subsequent acylation, affording triene 43, which in turn was submitted to RCM catalysed by second generation Hoveyda–Grubbs catalyst, forging the shagenes’ complex tricyclic core (44) in 86% yield after hydrolysis.
With the shagenes’ carbon framework successfully constructed, further functional group manipulations gave first intermediate 45, namely by dehydration with Burgess reagent to install the enone on the 5-membered ring, followed by isolated double bond isomerisation to iso-propenyl group on the cyclopropane ring with Crabtree's catalyst.41 Subsequent methyllithium 1,2-addition, followed by Dauben oxidative allylic rearrangement, and TBS group exchange to acetyl provided access to shagene B (46), while enone 1,2-reduction with DIBAL-H followed by methylation with MeI/NaH before the hydroxy-group protection exchange provided shagene A. Interestingly, optical rotation data provided evidence that the enantiomers of the shagenes were obtained in synthetic form. This synthesis serves as a highly interesting example for the construction of small rings based on the reaction of diazoketones with allenes.
Peyssonnosol
Peyssonosol (21) provides another unique carbocyclic system with a cyclopropane ring in the heart of the structure. This aglucone of peyssonnosides A and B was isolated by Kubanek and co-workers in 201942 as marine sulfated ditepenoid glucosides. This natural product features a 5/6/3/6-membered tetracyclic skeleton with an unusual pentasubstituted cyclopropane. Recently, Chesnokov and Gademann22 reported the successful total synthesis of this polycyclic terpenoid target (Scheme 2). The challenge of the cyclopropane was its liability to ionic or radical conditions during the course of the synthesis, which required careful orchestration of the sequential ring construction starting from a 5-membered cyclic precursor. | ||
| Scheme 2 Total synthesis of peyssonnosol by Chesnokov and Gademann. | ||
The literature known43 cyclic silyl enol ether 48 was reacted with enone 47 in Mukaiyama–Michael reaction under BF3·Et2O catalysis. Subsequent base-mediated condensation and cyclisation gave compound 50, finishing the Robinson annulation sequence. Diastereoselective Luche reduction set the stage for the key cyclopropane installation. Furukawa modification44 of the Simmons–Smith reaction performed with iodoform provided exclusively one product (51) in 72% yield with the desired configuration, furnishing construction of the third cycle of the peyssonnosol skeleton. We could rationalize these findings by the directing effect of the hydroxy group and the conformational properties of the substrate. Noteworthy, three stereocentres, two of which are quaternary, were installed in one chemical step.
The last 6-membered cycle was envisioned to be closed via RCM, which needed some further functional group manipulations in order to get the corresponding substrate. Thus, the iodine atom in 51 was substituted by a methallyl group via the Corey–House reaction45 (52), after preliminary TMS protection of the free hydroxy-group. The second olefin moiety was installed via Grieco dehydration protocol,46 employed on 3-hydroxypropyl side chain of 52. Ring-closing methatesis on the compound 52 provided the 5/6/3/6-teracyclic core of peyssonnosides in quantitative yield. The synthesis was finished by installation of the methyl group in place of secondary alcohol moiety via Ley–Griffith oxidation, Wittig methenylation, and hydrogenation over Rh/Al2O3, and tertiary alcohol by selective anaerobic Mukaiyama hydration.47 The peyssonnoside A synthesis is concise and highly efficient, providing peyssonnosol (21) in 21% yield after 13 steps.
Mitrephorone A
The diterpenoid mitrephorone A (24) from Bornean shrub Mitrephora glabra, isolated in 2015 by Oberlies and co-workers,48 contains a pentacyclic core decorated with oxetane ring. Moreover, the tricyclo[3.2.1.02,7]octane scaffold as part of the carbon skeleton of mitrophorone renders extra molecular complexity to the system. Further accentuating the chemical challenges, the presence of an oxetane, a cyclopropane, and the diketone render this target daunting. The first enantioselective total synthesis of (–)-mitrephorone A (24) was reported by Carreira and co-workers in 2018 (Scheme 3)25a and also by Magauer and co-workers (Chart 1).25c | ||
| Scheme 3 Total synthesis of (–)-mitrephorone A by Carreira and co-workers. | ||
The Carreira synthesis starts with the construction of the cage-like cyclopropane-containing tricyclooctane core 59. TADDOL-catalysed Diels–Alder reaction49 between Rawal's diene (55) and methacrolein followed by Wittig methenylation afforded cyclohexenone 57 in 70% and 88% ee. Further acylation of the ketone enolate with Mander's reagent and TBS protection of the product provided triene 58. Heating of the triene 58 in toluene at 190 °C gave 59 (after reduction and hydrolysis) as a single product, with the use of propylene oxide as a scavenger. This approach exemplifies a unique case of a cyclopropane synthesis via Diels–Alder reaction in complex molecule assembly.
The primary OH moiety in 59 was converted to the cyanide, followed by addition of penta-2,4-dien-2-yllithium to the carbonyl group of 60 in presence of Knochel's LaCl3·2LiCl reagent.50 The limited selectivity (dr 1.2:1) was rationalised by the poor induction mediated by the remote Me group breaking the mirror symmetry of the carbon framework in 60. Next, the resulting tertiary alcohol 61 was Si-protected and the nitrile group was reduced to the aldehyde 62 by DIBAL-H. Subsequent aldehyde alkynylation with lithium p-tolylsulfoxyacetylenide afforded the Diels–Alder substrate 63. Dess–Martin oxidation initiated the intramolecular Diels–Alder reaction, furnishing full construction of the carbon skeleton of mitrephorone A (24). It is noteworthy that the p-tolylsulfoxy-group and the resulting low-lying LUMO of the alkyne were crucial to the success of the Diels–Alder reaction, as the corresponding propyne substrate resulted only in poor yields.
The p-tolylsulfoxy group was then substituted by methyl with Me2CuLi to give 64. Hydrogenation of the isolated double bond in 64 over the Adams’ catalyst and subsequent Nagata 1,4-hydrocyanation of the α,β-enone moiety afforded ketonitrile 65. For the hydrolysis of the tertiary nitrile, the Ghaffar–Parkins catalyst51 was used with subsequent base hydrolysis of the forming amide. The resulting carboxylic acid group was methylated with TMSCHN2, followed by SeO2-mediated ketone α-oxidation to the α-diketone 66. During the last step of the synthesis the oxetane ring was oxidatively formed by treatment with Koser's reagent (PhI(OH)OTs). Overall, the synthesis features a series of highly sophisticated approaches based on deep mechanistic insight and state of the art methods and tactics.
Cyclobutastellettolide B
Another unusual polycyclic carbon framework can be found in cyclobutastellettolide B (7), marine derived terpenoid, isolated by Stonik and co-workers in 2019,52 and featuring 6/6/4-fused tricarbocyclic core with 5-membered lactone fused to the cyclobutane ring. The challenge associated with this small ring terpenoid relies in the sterically congested cyclobutane carbinol and its construction, with difficulties such as steric repulsion and poor orbital overlap. The total synthesis of this unusual and unique structure was recently reported by Huang, Yang and co-workers (Scheme 4).8 | ||
| Scheme 4 Total synthesis of (+)-cyclobutastellettolide B by Huang, Yang and co-workers. | ||
The synthesis starts from epoxide 67 available from geranyl acetate in 5 steps by literature known procedure.53 Cationic polyene cyclisation mediated by TiCl4 in dibromomethane54 successfully furnished the trans-decalin system of compound 68 in 86% yield on decagram scale. The obtained vinyl bromide 68 was converted to the corresponding allylic alcohol (69) by formylation of the preformed with t-BuLi vinyllithium species with DMF, followed by NaBH4 reduction. The allylic alcohol 69 was then treated with triethyl orthoacetate under heating and in presence of acid in order to perform Johnson–Claisen rearrangement55 to install carbethoxymethyl group. The rearrangement led to the axial product 70 in 84% yield. The exocyclic double bond was first epoxidised with m-CPBA, followed by Lewis acid catalysed rearrangement of the formed epoxide to the corresponding aldehyde, which in turn was converted to its Si-enol ether 71. The Si-protected enol ether 71 was then cyclopropanated from the α-face under Simmons–Smith conditions and the ring was opened with conc. aq. HCl solution to provide compound 72 with a correctly installed quaternary centre and an axial aldehyde moiety. It is worth noting that direct methylation of the aldehyde corresponding to 71 was not successful, giving predominantly O-alkylation byproducts, due to hindered equatorial attack trajectory for C-alkylation. On contrary, significantly different geometry of the Simmons–Smith cyclopropanation TS allowed for a clean and selective reaction via an equatorial approach.
The aldehyde moiety in 72 was then converted to the corresponding α-diketone (73) in 3 steps: (1) Wittig reaction with ethyl triphenylphosphonium bromide, (2) Os-catalysed dihydroxylation and (3) Swern oxidation. This set the stage for the last and key step of the synthesis – the Norrish–Yang photocyclisation56 between C(9) and C(14). Thus, irradiation of 73 in chloroform resulted in clean conversion to (+)-cyclobutastellettolide B (7) in 95% yield with successful installation of cyclobutane ring and subsequent formation of the 5-membered lactone ring. As a testament to this synthesis, the Norrish–Yang photocyclisation afforded clean formation of the bicyclic C–D motif in one step. The success of the transformation can be attributed to the rigid trans-decalin core and clever and controlled installation of the stereocentres previously in the sequence.
Canataxpropellane
Taxane diterpenoids are a medicinally important family of natural products exhibiting potent anticancer activity. To date more than 500 different taxanes have been isolated. In 2007 Kiyota and co-workers isolated canataxpropellane (19), one of the most complex natural products from taxane family.57 It comprises a hexacyclic 5/5/4/6/6/6 carbon framework with two propellane substructures in it. A fully substituted cyclobutane ring in the centre of the cage-like molecule significantly adds to the synthetic challenge. The synthesis of this complex natural product was reported by Gaich and co-workers in 2020 (Scheme 5).20 The synthetic strategy notes the central position of the cyclobutane ring, putting its formation in the very first steps, with subsequent decoration of the complex carbon framework. | ||
| Scheme 5 Total synthesis of canataxpropellane by Gaich and co-workers. | ||
The synthesis starts with Diels–Alder reaction between compound 75 and TBS protected compound 74. The endo-product 76 was isolated in 73% yield. In the next step the Diels–Alder adduct (76) was subjected to UV irradiation and underwent dearomative [2+2] cycloaddition in 73% yield to give 77. With this step, 5 out of 6 cycles of the target structure have been installed, including the octasubstituted cyclobutane ring. In order to install the last cycle, considerable modification of the former aromatic ring was needed. This sequence started with breaking the ether between C(2) and C(20) via retro-aldol reaction, translactonisation, and protection of the released hydroxy group at C(2) with a MOM-group to give 78. This compound was reduced and reoxidised to ketoaldehyde form and treated with t-BuOK in THF to restore the bond broken during the retro-aldol step. After this six step elaboration, the compound 79 was treated with singlet oxygen, and thus formed endo-peroxide was opened to result in the γ-hydroxyenone in the attached 6-membered ring. In order to invert the C(5) stereocentre, the hydroxy group was oxidised with IBX/DMSO and then the resulting carbonyl group was diastereoselectively reduced with tetramethylammonium triacetoxyborohydride to give 80. Subsequent protection of diol at C(5) and C(20) as benzaldehyde acetal set the stage for the formation of the last ring of canataxpropellane (19). This sequence started with 1,4-reduction of the enone by L-selectride, followed by triflation, and Pd-catalysed carbonylation to form the α,β-unsaturated methyl ester moiety (81). This in turn was reduced to the saturated methyl ester by Mg in MeOH. α-Methylation of the ester and conversion to aldehyde was achieved via two step sequence of LiAlH4 reduction and Swern oxidation.
The dialdehyde compound 82 was treated with low valent Ti to furnish pinacol coupling between the aldehyde groups, thus forming the last ring of the canataxpropellane skeleton. The pinacol coupling gave the desired anti-configuration of the diol system at C(9) and C(10). The following 5 steps served to selectively decorate the hydroxy groups at C(10) and C(2) with acetyl groups.
With this the synthesis of the most complex taxane, canataxpropellane (19), was successfully achieved. It is interesting to note that there are only 5 C–C bond forming steps in this synthesis with the majority of steps being reduction/oxidation or protection/deprotection, which can be attributed to high level of functionalisation of the natural product. This route truly constitutes a remarkable and spectacular achievement in terpenoid synthesis.
Harziane
In 2014, unusual diterpenoids were isolated from a Trichoderma symbiont of Taxus baccata by Ouazzani and co-workers.58 The tetracyclic diterpenoid harziane (20) stood out, featuring a 6/5/7/4-fused carbon skeleton. The structure also harbors six contiguous stereocenters, two of which are quaternary, which render the target challenging. The total synthesis of this natural product was reported in 2019 by Hönig and Carreira (Scheme 6).21 The synthesis features an unusual Au-catalysed cycloisomerisation as a key step. | ||
| Scheme 6 Total synthesis of harziane by Hönig and Carreira. | ||
The synthesis starts from literature known compound 84, which was first converted to cyclopentane 85 by ester α-alkylation and subsequent Pd-catalysed cycloisomerisation59 to give 85 in 77% overall yield. The ester group was methylated with methyl magnesium bromide via a Weinreb amide to give the corresponding methyl ketone which was further protected as 1,3-dioxolane (86). The diene system in 86 was first doubly hydroxylated through hydroboration; the primary alcohol was protected as a pivalate, and the secondary OH group was oxidised to the methyl ketone 87. This ketone was then submitted to Tebbe olefination with the corresponding Petasis reagent60 to install cyclopropylene moiety. The protected primary alcohol side chain was then deprotected and after Ley–Griffith oxidation converted to ethynyl group with the Bestmann–Ohira regent to give enyne 88, ready for the key cycloisomerisation step. The key step was based on a Gagné report of an interesting tethered enyne cycloisomerisation leading to a 6/4-fused bicyclic diene.61 Thus, treatment with a triphenylphosphine gold catalyst afforded a clean diastereoselective (dr >11:1) conversion to the tricyclic 5/6/4 system of the compound 89 in 87% yield.
The intermediate 89 was then selectively hydroborated with ThxBH2, followed by Ley–Griffith oxidation to give enone, which was then treated with the Nagata's reagent (Et2AlCN) to selectively obtain ketonitrile 90. After triflation and Negishi coupling with dimethylzinc, the ketone was converted to the corresponding methylated olefin, followed by deprotection of 1,3-dioxolane and conversion of the formed methylketone to the corresponding TBS-protected enol ether 91. The nitrile in 91 was reduced with DIBAL-H to the corresponding aldimine, which could be isolated after aqueous workup and chromatography. Exposure to silica led to the intramolecular aldol reaction. The formed enone was then 1,4-methylated under Yamamoto's conditions62 to give 92. Next, several steps were carried out to enlarge the central six membered ring by one C atom. Oxidative cleavage under Ru-catalysis with subsequent LiHMDS-induced intramolecular aldol cyclisation afforded 93 after Wittig methenylation. In the finishing phase of the synthesis, both O atoms were synthetically deleted (reduction, then Barton–McCombie sequence). Synthesis of both tertiary alcohols and evaluation of NMR data led to a structural revision of this stereogenic center, and the correct configuration was selectively obtained via Mukaiyama hydration conditions affording harziane (20). The elegant use of modern Au-mediated ring formation reactions constitutes a spectacular example of the power of these transformations.
Atlanticone C
Atlanicone C (6) is a member of the protoilludane family sesquiterpenoids featuring 5/6/4-fused tricyclic scaffold.63 A very elegant approach to this target was recently reported by Bach and co-workers (Scheme 7), utilising a photochemical reaction cascade.7 | ||
| Scheme 7 Total synthesis of atlanticone C by Bach and co-workers. | ||
The synthesis starts with the indanone 96, easily available by alkylation sequence from indanone 95, which in turn can be prepared by tandem Fries rearrangement and Friedel–Crafts cyclisation in one-pot fashion.64 The indanone 96 upon irradiation with near UV light undergoes a sequence of [2+2] reaction, electrocyclic ring opening, and 4π-electrocyclisation to result in the drastically transformed compound 97 in 60% yield with carbon skeleton of atlanticone C (6) constructed.
Further functional decoration sequence includes hydrogenation of the isolated double bond, conversion of the enone to the corresponding tosylhydrazone 98, which was reduced under Kabalka's conditions65 with double bond transposition, allowing for the stereoselective installation of the fused methine C–H group. A clever cleavage of the methylene protection via lithiation and quenching with BF(OMe)2, followed by oxidative removal led to the diol system (99).66 The synthesis end stage included Swern oxidation of the primary OH group to the aldehyde, dehydration of tertiary alcohol and reduction of the CHO group back to the primary alcohol (100). Further installation of the carbonyl group by chromium trioxide oxidation with temporary protection of the primary alcohol as acetate resulted in atlanticone C (6). This synthesis is noteworthy for its rapid and highly elegant construction of the complex carbocyclic framework via photochemical conditions.
Plumisclerin A
In 2010, Reyes and co-workers isolated a new member of the xenicane diterpenoid family from the soft coral Plumigorgia terminosclera.67 The natural product named plumisclerin A (3) features a 5/4/6-fused tricyclic ring system with another fused dihydropyran ring. The presence of a central cyclobutane ring further accentuates the complexity of this target. The synthesis of this structurally interesting natural product was first reported 2018 by Yao and co-workers (Scheme 8).4 | ||
| Scheme 8 Total synthesis of (+)-plumesclerin A by Yao and co-workers. | ||
The enantioselective 1,4-addition of homoallylated dibenzyl malonate (102) to cyclopentenone under Shibasaki's conditions opened the synthesis.68 Then, the vinyl part of homoallyl sidechain was cleaved by ozonolysis, releasing an aldehyde, which upon acidic treatment underwent aldol condensation in 63% yield to give bicyclic compound 104. Two ester groups were then reduced with LiAlH4 and protected as a 1,3-dioxane, followed by oxidation and hydrogenation of the resulting enone double bond affording ketone 105. This ketone was alkylated with methyl bromoacetate (≥ 106), subsequently triflated to the vinyl triflate, which in turn was further carbonylated under Pd catalysis in MeOH to provide installation of carbmethoxy group in 107.
The diol protection was removed under acidic conditions. One of the OH groups was selectively protected with TBSCl/Im-H, and the other one was converted to the corresponding aldehyde with PCC, affording the substrate (108) for the key step. Then the cyclobutane ring was formed via SmI2-mediated ring closure through Giese reaction of the ketyl radical of the aldehyde to the unsaturated ester counterpart,69 followed by TBS protection of newly formed alcohol 109 (selectivity 3.3:1 dr).
Further, the compound 109 was carbmethoxylated α to the less sterically hindered ester. The newly formed malonate moiety was reduced with DIBAL-H to the corresponding diol, which upon treatment with camphorsulfonic acid underwent cyclisation to 6-membered lactone, affording 110 after TES protection. Then, the lactone was converted to the corresponding lactol with DIBAL-H and protected with acetate. The primary alcohol in a sidechain was then oxidised by Swern conditions to the corresponding aldehyde 111. α-Selenylation of the newly formed aldehyde, catalysed by L-prolinamide,70 successfully installed the double bond, conjugated to the aldehyde (112). The finishing steps included addition of the iso-amyl Grignard reagent to the aldehyde with subsequent oxidation to ketone, TBS deprotection, and conversion of all the hydroxy groups to acetates. Thus, the total synthesis of (+)-plumisclerin A (3) was successfully accomplished. The fascinating use of an intramolecular Giese addition furnishing the cyclobutane ring is a true masterpiece of modern radical chemistry in terpene total synthesis.
Dendrowardol C
Dendrowardol C (23) was isolated in 2013 from an orchid Dendrobium wardianum, endemic to southern China and Southeast Asia.71 This natural product features an interesting and unique 6/5/5/4-fused tetracyclic ring system, decorated with several hydroxy-groups. Wolleb and Carreira reported the first total synthesis of this target in 2017 (Scheme 9).24 In their synthesis, late stage instalment of the cyclobutane ring was key to a successful preparation of the natural product. | ||
| Scheme 9 Total synthesis of dendrowardol C by Wolleb and Carreira. | ||
The synthesis starts with alkylation of (R)-carvone with ethyl bromoacetate to give 114. This compound was further sequentially reduced by L-selectride and LiAlH4, and then oxidised back to the dicarbonyl stage with the Swern method to give compound 115. This compound when treated with methanolic sodium methoxide solution cyclised to the bridged bicyclic ketol 116. This compound was then oxidised with Dess–Martin periodinane, and the more available bridgehead carbonyl group was then homologated by Wittig olefination with MOMPPh3Cl to give 117 after hydrolysis. Selective addition of vinyllithium to the aldehyde (118) followed by a diastereoslective directed epoxidation of the newly formed allylic alcohol under Sharpless conditions with VO(acac)2/t-BuOOH system afforded compound 119 after subsequent TMS protection.
The compound 119 upon treatment with LDA underwent proximity-induced epoxide opening via the Li enolate forming the five membered ring in 53% yield (120). Triflation of the primary alcohol in the sidechain gave 121 in 65% yield. The key cyclobutane cycle formation was performed with Li naphtalenide in 2-MeTHF/benzene mixture at 10 °C, affording the product 122 in 46% yield.72 Interestingly, the course of this reaction is not fully understood and the authors provided both putative anionic and radical-anionic pathways in order to explain the outcome. The synthesis of (+)-dendrowardol C (23) was finished by asymmetric Co-catalysed hydroboration with HBpin,73 followed by oxidation to the corresponding primary alcohol. This approach is noteworthy for its brevity in establishing molecular complexity, and the notion that sometimes interesting reactions, such as the cyclobutane synthesis (121 → 122), can be rationalised by a multitude of different mechanistic pathways.
Aplydactone
Aplydactone (22) was isolated by Stonik and co-workers from the sea hare Aplysia dactylomela and features a unique 6/6/4/4-ring architecture with two fused cyclobutane rings.74 This natural product also contains secondary Br substituent on the 6-membered ring, which renders this target extremely challenging to organic synthesis. To date, there are three successful total syntheses reported: by Burns and co-workers in 2016,23a by Meier and Trauner in 2016,23b and by Zhang and co-workers in 201723c (Scheme 10). We will review all of them to compare different approaches to such a unique carbon framework. | ||
| Scheme 10 Total syntheses of aplydactone: (a) by Burns and co-workers; (b) by Meier and Trauner; (c) by Zhang and co-workers. | ||
The synthesis by Burns and co-workers starts with terminal SeO2 allylic oxidation of geranyl acetate (123), followed by enantioselective bromochlorination, developed previously in the same group, to give 124 in 66% yield and 94% ee. This compound was partially reduced to remove the free hydroxy-group by a sequence of triflation and treatment with L-selectride, followed by acetylation to restore the original acetate group (125). Then, the compound 125 was treated with K2CO3 in HFIP. Highly polar media induced ionisation of the tertiary chloride facilitated the desired cyclisation with the olefin moiety to give bromocyclohexanol 126 in 54% yield in an enantiospecific manner. Then, the tertiary alcohol was removed by chlorination with SOCl2 and elimination with Et3N, followed by oxidative RuCl3-mediated cleavage of the intermediate exo-methylene group, giving compound 127. This compound was in situ converted to the corresponding enone and reacted with isoprene under Me2AlCl catalysis to give the Diels–Alder product 128 in 52% yield along with 12% of its regioisomer. The carbonyl group in 128 was then methenylated under Lombardo type conditions with Mg/TiCl4/DCM system.75 Then, SeO2 allylic oxidation with subsequent IBX oxidation afforded the natural product (+)-dactylone (129). Near UV light irradiation of (+)-dactylone afforded (+)-aplydactone (22) in 98% yield through photoinduced [2+2] cycloaddition, demonstrating that dactylone could be a biosynthetic precursor of aplydactone.
The other two syntheses employ very different, but similar to each other retrosynthetic logic. The synthesis by Meier and Trauner starts with the compound 130, which was converted into 133 after a series of 11 steps. The compound 133 was in turn irradiated by UV light to induce photo-[2+2] cycloaddition between olefin and enone counterparts to give tricyclic ketone 134 in 55% yield. Then, this 6/4/5 tricyclic compound (134) was subjected to Wolff rearrangement-based ring contraction in order to generate the fused 6/4/4 system. The ring contraction worked in 47% yield over two steps via diazo compound formation (135). In the following 6 steps, the ester 135 was α-methylated (136) and later converted to the Weinreb amide, the protected primary alcohol was converted into iodide, and the secondary alcohol was reprotected with TIPS group (137). The final 5-membered cycle was formed by treatment of 137 with t-BuLi, inducing Li/I exchange with subsequent intramolecular trapping by the Weinreb amide, affording the full aplydactone carbon skeleton in 42% yield (138), subsequent radical substitution of the secondary alcohol to the secondary bromide afforded the target aplydactone (22).
Zhang and co-workers’ synthesis of aplydactone (22) employs the same ring forming strategy as Trauner's route, but does it in half the number of steps. The synthesis starts with addition of 1,3-dioxolan-2-ylethyl Grignard reagent to the 139, followed by Stork–Danheiser transposition and dioxolane deprotection upon acidification to give keto aldehyde which was then prenylated at the aldehyde centre under Barbier conditions, affording 140 in 34% overall yield. UV light irradiation of 140 induced photo-[2+2] cycloaddition to give 6/4/5-fused tricyclic 141, similar to 134. This compound was also subjected to ring contraction by Wolff rearrangement, followed by α-methylation of the forming ester to give 142. The secondary OH group in 142 was then converted to vinyl bromide by Dess–Martin oxidation, formation of the corresponding tosylhydrazone and subsequent bromination with NBS76 in overall 50% yield (143). The last cycle was closed via Rh-catalysed carbene insertion into C–H bond: The ester was hydrolysed with LiOH, followed by generation of the terminal diazoketone via the corresponding acyl chloride. This diazo compound underwent unselective carbenoid insertion (competing insertions into C(6)-H and closer in space C(12)-H bonds) under Rh2(tfa)4 catalysis to give 144 in 34% yield over two steps along with its isomer, which was the major product of insertion. Aplydactone (22) was then synthesised by an interesting HAT-based reduction of the bromoolefin moiety in 144 with Mn(dpm)3 and Ph(i-PrO)SiH2/t-BuOOH.77 In summary, the different and complementary approaches nicely illustrate the power of photochemical transformations in complex molecule assembly and even might lend to hypotheses concerning the biogenesis of such compounds.
Conclusions
In summary, we have discussed recent highlights in the field of small ring containing polycyclic terpenoids. All these examples highlight ingenious strategies combined with state-of-the-art methods. Each of the natural products discussed poses unique challenges that were met by sophisticated approaches carefully orchestrating the sequence of reactions and culminating in truly remarkable masterpieces of molecular art in the form of terpene natural products.Author contributions
Gleb A. Chesnokov: conceptualisation, visualisation, writing – original draft, writing – review & editing; Karl Gademann: conceptualisation, funding acquisition, project administration, supervision, writing – review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Support by the Swiss National Science Foundation (182043) is gratefully acknowledged.References
- (a) T. Bach and J. P. Hehn, Angew. Chem., Int. Ed., 2011, 50, 1000–1045 CrossRef CAS PubMed; (b) Z. G. Brill, M. L. Condakes, C. P. Ting and T. J. Maimone, Chem. Rev., 2017, 117, 11753–11795 CrossRef CAS PubMed; (c) C. Ebner and E. M. Carreira, Chem. Rev., 2017, 117, 11651–11679 CrossRef CAS PubMed; (d) K. Hung, X. Hu and T. J. Maimone, Nat. Prod. Rep., 2018, 35, 174–202 RSC; (e) D. J. Jansen and R. A. Shenvi, Future Med. Chem., 2014, 6, 1127–1148 CrossRef CAS PubMed; (f) M. D. Kärkäs, J. A. Porco and C. R. J. Stephenson, Chem. Rev., 2016, 116, 9683–9747 CrossRef PubMed; (g) J. Li, K. Gao, M. Bian and H. Ding, Org. Chem. Front., 2020, 7, 136–154 RSC; (h) T. Qiao and G. Liang, Sci. China: Chem., 2016, 59, 1142–1174 CrossRef CAS; (i) D. Urabe, T. Asaba and M. Inoue, Chem. Rev., 2015, 115, 9207–9231 CrossRef CAS PubMed; (j) Y. Zou and A. B. Smith, J. Antibiot., 2018, 71, 185–204 CrossRef CAS PubMed.
- M. T. Hovey, D. T. Cohen, D. M. Walden, P. H. Y. Cheong and K. A. Scheidt, Angew. Chem., Int. Ed., 2017, 56, 9864–9867 CrossRef CAS PubMed.
- C. Tsukano, R. Yagita, T. Heike, T. A. Mohammed, K. Nishibayashi, K. Irie and Y. Takemoto, Angew. Chem., Int. Ed., 2021, 60, 23106–23111 CrossRef CAS PubMed.
- M. Gao, Y.-C. Wang, K.-R. Yang, W. He, X.-L. Yang and Z.-J. Yao, Angew. Chem., Int. Ed., 2018, 57, 13313–13318 CrossRef CAS PubMed.
- (a) J. Xuan, Z. Liu, A. Zhu, P. Rao, L. Yu and H. Ding, Angew. Chem., Int. Ed., 2017, 56, 8898–8901 CrossRef CAS PubMed; (b) P. Yuan, C. K. G. Gerlinger, J. Herberger and T. Gaich, J. Am. Chem. Soc., 2021, 143, 11934–11938 CrossRef CAS PubMed.
- S. Pan, J. Xuan, B. Gao, A. Zhu and H. Ding, Angew. Chem., Int. Ed., 2015, 54, 6905–6908 CrossRef CAS PubMed.
- A. Zech, C. Jandl and T. Bach, Angew. Chem., Int. Ed., 2019, 58, 14629–14632 CrossRef CAS PubMed.
- Z. Zhang, S. Chen, F. Tang, K. Guo, X.-T. Liang, J. Huang and Z. Yang, J. Am. Chem. Soc., 2021, 143, 18287–18293 CrossRef CAS PubMed.
- H. Abe, T. Morishita, T. Yoshie, K. Long, T. Kobayashi and H. Ito, Angew. Chem., Int. Ed., 2016, 55, 3795–3798 CrossRef CAS PubMed.
- (a) S.-H. Hou, Y.-Q. Tu, S.-H. Wang, C.-C. Xi, F.-M. Zhang, S.-H. Wang, Y.-T. Li and L. Liu, Angew. Chem., Int. Ed., 2016, 55, 4456–4460 CrossRef CAS PubMed; (b) P. Hu, H. M. Chi, K. C. DeBacker, X. Gong, J. H. Keim, I. T. Hsu and S. A. Snyder, Nature, 2019, 569, 703–707 CrossRef CAS PubMed; (c) B. Xu, W. Xun, S. Su and H. Zhai, Angew. Chem., Int. Ed., 2020, 59, 16475–16479 CrossRef CAS PubMed.
- M. J. Classen, M. N. A. Böcker, R. Roth, W. M. Amberg and E. M. Carreira, J. Am. Chem. Soc., 2021, 143, 8261–8265 CrossRef CAS PubMed.
- N. Zhao, S. Yin, S. Xie, H. Yan, P. Ren, G. Chen, F. Chen and J. Xu, Angew. Chem., Int. Ed., 2018, 57, 3386–3390 CrossRef CAS PubMed.
- (a) A. C. Wright and B. M. Stoltz, Chem. Sci., 2019, 10, 10562–10565 RSC; (b) A. Hirose, A. Watanabe, K. Ogino, M. Nagatomo and M. Inoue, J. Am. Chem. Soc., 2021, 143, 12387–12396 CrossRef CAS PubMed.
- N. J. Hafeman, S. A. Loskot, C. E. Reimann, B. P. Pritchett, S. C. Virgil and B. M. Stoltz, J. Am. Chem. Soc., 2020, 142, 8585–8590 CrossRef CAS PubMed.
- (a) T. Yu, Y. Sun, C. Tu, T. Chen, S. Fu and B. Liu, Chem. Sci., 2020, 11, 7177–7181 RSC; (b) C. Cui, B. G. Dwyer, C. Liu, D. Abegg, Z.-J. Cai, D. G. Hoch, X. Yin, N. Qiu, J.-Q. Liu, A. Adibekian and M. Dai, J. Am. Chem. Soc., 2021, 143, 4379–4386 CrossRef CAS PubMed.
- S. Kawamura, H. Chu, J. Felding and P. S. Baran, Nature, 2016, 532, 90–93 CrossRef CAS PubMed.
- Y. Li and M. Dai, Angew. Chem., Int. Ed., 2017, 56, 11624–11627 CrossRef CAS PubMed.
- H.-D. Hao and D. Trauner, J. Am. Chem. Soc., 2017, 139, 4117–4122 CrossRef CAS PubMed.
- (a) P.-P. Zhang, Z.-M. Yan, Y.-H. Li, J.-X. Gong and Z. Yang, J. Am. Chem. Soc., 2017, 139, 13989–13992 CrossRef CAS PubMed; (b) C. He, J. Xuan, P. Rao, P.-P. Xie, X. Hong, X. Lin and H. Ding, Angew. Chem., Int. Ed., 2019, 58, 5100–5104 CrossRef CAS PubMed.
- F. Schneider, K. Samarin, S. Zanella and T. Gaich, Science, 2020, 367, 676–681 CrossRef CAS PubMed.
- M. Hönig and E. M. Carreira, Angew. Chem., Int. Ed., 2020, 59, 1192–1196 CrossRef PubMed.
- G. A. Chesnokov and K. Gademann, J. Am. Chem. Soc., 2021, 143, 14083–14088 CrossRef CAS PubMed.
- (a) A. J. Burckle, V. H. Vasilev and N. Z. Burns, Angew. Chem., Int. Ed., 2016, 55, 11476–11479 CrossRef CAS PubMed; (b) R. Meier and D. Trauner, Angew. Chem., Int. Ed., 2016, 55, 11251–11255 CrossRef CAS PubMed; (c) C. Liu, R. Chen, Y. Shen, Z. Liang, Y. Hua and Y. Zhang, Angew. Chem., Int. Ed., 2017, 56, 8187–8190 CrossRef CAS PubMed.
- H. Wolleb and E. M. Carreira, Angew. Chem., Int. Ed., 2017, 56, 10890–10893 CrossRef CAS PubMed.
- (a) M. J. R. Richter, M. Schneider, M. Brandstätter, S. Krautwald and E. M. Carreira, J. Am. Chem. Soc., 2018, 140, 16704–16710 CrossRef CAS PubMed; erratum, J. Am. Chem. Soc., 2021, 143, 9694 Search PubMed; (b) M. Schneider, M. J. R. Richter and E. M. Carreira, J. Am. Chem. Soc., 2020, 142, 17802–17809 CrossRef CAS PubMed; (c) L. A. Wein, K. Wurst, P. Angyal, L. Weisheit and T. Magauer, J. Am. Chem. Soc., 2019, 141(50), 19589–19593 CrossRef CAS PubMed.
- N. Rauscher, L. Næsborg, C. Jandl and T. Bach, Angew. Chem., Int. Ed., 2021, 60, 24039–24042 CrossRef CAS PubMed.
- R. Liffert, A. Linden and K. Gademann, J. Am. Chem. Soc., 2017, 139, 16096–16099 CrossRef CAS PubMed.
- Y. Wang, A. Jäger, M. Gruner, T. Lübken and P. Metz, Angew. Chem., Int. Ed., 2017, 56, 15861–15865 CrossRef CAS PubMed.
- (a) L. Min, X. Lin and C.-C. Li, J. Am. Chem. Soc., 2019, 141, 15773–15778 CrossRef CAS PubMed; (b) X. Yu, L. Xiao, Z. Wang and T. Luo, J. Am. Chem. Soc., 2019, 141, 3440–3443 CrossRef CAS PubMed.
- (a) H.-J. Zhang, L. Hu, Z. Ma, R. Li, Z. Zhang, C. Tao, B. Cheng, Y. Li, H. Wang and H. Zhai, Angew. Chem., Int. Ed., 2016, 55, 11638–11641 CrossRef CAS PubMed; (b) Q. Ao, H.-J. Zhang, J. Zheng, X. Chen and H. Zhai, Angew. Chem., Int. Ed., 2021, 60, 21267–21271 CrossRef CAS PubMed.
- J. Gao, P. Rao, K. Xu, S. Wang, Y. Wu, C. He and H. Ding, J. Am. Chem. Soc., 2020, 142, 4592–4597 CrossRef CAS PubMed.
- Z. Ren, Z. Sun, Y. Li, X. Fan, M. Dai, Y. Wang and X. Hu, Angew. Chem., Int. Ed., 2021, 60, 18572–18576 CrossRef CAS PubMed.
- L. Xu, C. Wang, Z. Gao and Y.-M. Zhao, J. Am. Chem. Soc., 2018, 140, 5653–5658 CrossRef CAS PubMed.
- (a) H. Zhang, H. He and S. Gao, Angew. Chem., Int. Ed., 2020, 59, 20417–20422 CrossRef CAS PubMed; (b) M. Haider, G. Sennari, A. Eggert and R. Sarpong, J. Am. Chem. Soc., 2021, 143, 2710–2715 CrossRef CAS PubMed.
- (a) Z. Huang, J. Huang, Y. Qu, W. Zhang, J. Gong and Z. Yang, Angew. Chem., Int. Ed., 2018, 57, 8744–8748 CrossRef CAS PubMed; (b) Y. Zhao, J. Hu, R. Chen, F. Xiong, H. Xie and H. Ding, J. Am. Chem. Soc., 2022, 144, 2495–2500 CrossRef CAS PubMed.
- (a) H. Lee, T. Kang and H.-Y. Lee, Angew. Chem., Int. Ed., 2017, 56, 8254–8257 CrossRef CAS PubMed; (b) Y. Qu, Z. Wang, Z. Zhang, W. Zhang, J. Huang and Z. Yang, J. Am. Chem. Soc., 2020, 142, 6511–6515 CrossRef CAS PubMed; (c) C. Peng, P. Arya, Z. Zhou and S. A. Snyder, Angew. Chem., Int. Ed., 2020, 59, 13521–13525 CrossRef CAS PubMed; (d) L.-C. Rosenbaum, M. Häfner and T. Gaich, Angew. Chem., Int. Ed., 2021, 60, 2939–2942 CrossRef CAS PubMed.
- J. L. von Salm, N. G. Wilson, B. A. Vesely, D. E. Kyle, J. Cuce and B. J. Baker, Org. Lett., 2014, 16, 2630–2633 CrossRef CAS PubMed.
- M. Ostermeier, B. Brunner, C. Korff and G. Helmchen, Eur. J. Org. Chem., 2003, 3453–3459 CrossRef CAS.
- S. K. Murphy and V. M. Dong, J. Am. Chem. Soc., 2013, 135, 5553–5556 CrossRef CAS PubMed.
- B. Bolte, Y. Odabachian and F. Gagosz, J. Am. Chem. Soc., 2010, 132, 7294–7296 CrossRef CAS PubMed.
- B. Wüstenberg and A. Pfaltz, Adv. Synth. Catal., 2008, 350, 174–178 CrossRef.
- B. Khatri Chhetri, S. Lavoie, A. M. Sweeney-Jones, N. Mojib, V. Raghavan, K. Gagaring, B. Dale, C. W. McNamara, K. Soapi, C. L. Quave, P. L. Polavarapu and J. Kubanek, J. Org. Chem., 2019, 84, 8531–8541 CrossRef CAS PubMed.
- E. Piers and J. Renaud, J. Org. Chem., 1993, 58, 11–13 CrossRef CAS.
- J. Furukawa, N. Kawabata and J. Nishimura, Tetrahedron Lett., 1968, 9, 3495–3498 CrossRef.
- E. J. Corey and G. H. Posner, J. Am. Chem. Soc., 1967, 89, 3911–3912 CrossRef CAS.
- P. A. Grieco, S. Gilman and M. Nishizawa, J. Org. Chem., 1976, 41, 1485–1486 CrossRef CAS.
- A. Bhunia, K. Bergander, C. G. Daniliuc and A. Studer, Angew. Chem., Int. Ed., 2021, 60, 8313–8320 CrossRef CAS PubMed.
- C. Li, D. Lee, T. N. Graf, S. S. Phifer, Y. Nakanishi, J. P. Burgess, S. Riswan, F. M. Setyowati, A. M. Saribi, D. D. Soejarto, N. R. Farnsworth, J. O. Falkinham, D. J. Kroll, A. D. Kinghorn, M. C. Wani and N. H. Oberlies, Org. Lett., 2005, 7, 5709–5712 CrossRef CAS PubMed.
- A. N. Thadani, A. R. Stankovic and V. H. Rawal, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 5846 CrossRef CAS PubMed.
- A. Krasovskiy, F. Kopp and P. Knochel, Angew. Chem., Int. Ed., 2006, 45, 497–500 CrossRef CAS PubMed.
- (a) T. Ghaffar and A. W. Parkins, Tetrahedron Lett., 1995, 36, 8657–8660 CrossRef CAS; (b) X.-b. Jiang, A. J. Minnaard, B. L. Feringa and J. G. de Vries, J. Org. Chem., 2004, 69, 2327–2331 CrossRef CAS PubMed.
- S. A. Kolesnikova, E. G. Lyakhova, A. I. Kalinovsky, D. V. Berdyshev, E. A. Pislyagin, R. S. Popov, B. B. Grebnev, T. N. Makarieva, C. V. Minh and V. A. Stonik, J. Nat. Prod., 2019, 82, 3196–3200 CrossRef CAS PubMed.
- E. J. Corey, M. C. Noe and S. Wen-Chung, Tetrahedron Lett., 1993, 34, 5995–5998 CrossRef CAS.
- X.-T. Liang, J.-H. Chen and Z. Yang, J. Am. Chem. Soc., 2020, 142, 8116–8121 CrossRef CAS PubMed.
- W. S. Johnson, L. Werthemann, W. R. Bartlett, T. J. Brocksom, T.-T. Li, D. J. Faulkner and M. R. Petersen, J. Am. Chem. Soc., 1970, 92, 741–743 CrossRef CAS.
- S. Chen, Z. Zhang, C. Jiang, C. Zhao, H. Luo, J. Huang and Z. Yang, ACS Omega, 2021, 6, 18848–18859 CrossRef CAS PubMed.
- C.-H. Huo, X.-H. Su, Y.-F. Wang, X.-P. Zhang, Q.-W. Shi and H. Kiyota, Tetrahedron Lett., 2007, 48, 2721–2724 CrossRef CAS.
- E. Adelin, C. Servy, M.-T. Martin, G. Arcile, B. I. Iorga, P. Retailleau, M. Bonfill and J. Ouazzani, Phytochemistry, 2014, 97, 55–61 CrossRef CAS PubMed.
- B. M. Trost, G. J. Tanoury, M. Lautens, C. Chan and D. T. MacPherson, J. Am. Chem. Soc., 1994, 116, 4255–4267 CrossRef CAS.
- N. A. Petasis and E. I. Bzowej, Tetrahedron Lett., 1993, 34, 943–946 CrossRef CAS.
- H. Zheng, R. J. Felix and M. R. Gagné, Org. Lett., 2014, 16, 2272–2275 CrossRef CAS PubMed.
- Y. Yamamoto and K. Maruyama, J. Am. Chem. Soc., 1978, 100, 3240–3241 CrossRef CAS.
- M. Clericuzio, M. Mella, L. Toma, P. V. Finzi and G. Vidari, Eur. J. Org. Chem., 2002, 988–994 CrossRef CAS.
- W. D. Crow, U. Engkaninan-Low and Y. T. Pang, Aust. J. Chem., 1984, 37, 1915–1924 CrossRef CAS.
- M. L. Shrestha, W. Qi and M. C. McIntosh, J. Org. Chem., 2017, 82, 8359–8370 CrossRef CAS PubMed.
- (a) R. W. Hoffmann, G. Feussner, H.-J. Zeiss and S. Schulz, J. Organomet. Chem., 1980, 187, 321–329 CrossRef CAS; (b) K. Fujitta and M. Schlosser, Helv. Chim. Acta, 1982, 65, 1258–1263 CrossRef.
- M. J. Martín, R. Fernández, A. Francesch, P. Amade, S. S. de Matos-Pita, F. Reyes and C. Cuevas, Org. Lett., 2010, 12, 912–914 CrossRef PubMed.
- (a) S. Matsunaga, J. Das, J. Roels, E. M. Vogl, N. Yamamoto, T. Iida, K. Yamaguchi and M. Shibasaki, J. Am. Chem. Soc., 2000, 122, 2252–2260 CrossRef CAS; (b) R. Takita, T. Ohshima and M. Shibasaki, Tetrahedron Lett., 2002, 43, 4661–4665 CrossRef CAS.
- J.-P. Chen, W. He, Z.-Y. Yang and Z.-J. Yao, Org. Lett., 2015, 17, 3379–3381 CrossRef CAS PubMed.
- J. Wang, H. Li, Y. Mei, B. Lou, D. Xu, D. Xie, H. Guo and W. Wang, J. Org. Chem., 2005, 70, 5678–5687 CrossRef CAS PubMed.
- W.-W. Fan, F.-Q. Xu, F.-W. Dong, X.-N. Li, Y. Li, Y.-Q. Liu, J. Zhou and J.-M. Hu, Nat. Prod. Bioprospect., 2013, 3, 89–92 CrossRef CAS.
- E. Alonso, D. J. Ramón and M. Yus, Tetrahedron, 1996, 52, 14341–14348 CrossRef CAS.
- L. Zhang, Z. Zuo, X. Wan and Z. Huang, J. Am. Chem. Soc., 2014, 136, 15501–15504 CrossRef CAS PubMed.
- S. N. Fedorov, O. S. Radchenko, L. K. Shubina, A. I. Kalinovsky, A. V. Gerasimenko, D. Y. Popov and V. A. Stonik, J. Am. Chem. Soc., 2001, 123, 504–505 CrossRef CAS PubMed.
- T.-H. Yan, C.-C. Tsai, C.-T. Chien, C.-C. Cho and P.-C. Huang, Org. Lett., 2004, 6, 4961–4963 CrossRef CAS PubMed.
- D. P. Ojha and K. R. Prabhu, Org. Lett., 2015, 17, 18–21 CrossRef CAS PubMed.
- K. Iwasaki, K. K. Wan, A. Oppedisano, S. W. M. Crossley and R. A. Shenvi, J. Am. Chem. Soc., 2014, 136, 1300–1303 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See https://doi.org/10.1039/d2cc00926a |
| This journal is © The Royal Society of Chemistry 2022 |