
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceStraightforward synthesis of bench-stable heteroatom-centered difluoromethylated entities via controlled nucleophilic transfer from activated TMSCHF2†
Margherita
Miele
a,
Laura
Castoldi
b,
Xenia
Simeone
a,
Wolfgang
Holzer
a and
Vittorio
Pace
 *ac
*ac
aUniversity of Vienna - Department of Pharmaceutical Chemistry, Althanstrasse, 14 1090 Vienna, Austria. E-mail: vittorio.pace@univie.ac.at; Web: https://drugsynthesis.univie.ac.at/
bUniversity of Milano - Department of Pharmaceutical Sciences, Via Golgi 19, 20133 Milano, Italy
cUniversity of Torino - Department of Chemistry, Via Giuria 7, 10125 Torino, Italy
First published on 5th April 2022
Abstract
The commercially available and experimentally convenient (bp 65 °C) difluoromethyltrimethylsilane (TMSCHF2) is proposed as a valuable difluoromethylating transfer reagent for delivering the CHF2 moiety to various heteroatom-based electrophiles. Upon activation with an alkoxide, a conceptually intuitive nucleophilic displacement directly furnishes in high yields the bench-stable analogues.
Among the techniques enabling the modulation of pivotal physical–chemical properties of organic arrays, the introduction of fluorine atoms has become a powerful tool, nowadays thoroughly applied in chemistry.1 Synthetic chemists tackling the challenge of embodying fluorine into molecules quickly recognized that well-established methodologies in classical halogen chemistry could not be validated for this member of the series.2 Ideally, the use of fluorinated carbanion-like entities would enable – upon a conceptually intuitive nucleophile-electrophile logic – the forging of a new carbon–carbon bond presenting the exact and desired fluorination degree of the targeted compound (Scheme 1).3 However, F-containing nucleophiles are notoriously reluctant species mainly due to their inherent limited chemical integrity, which for a long time eclipsed their employment in synthesis.2a,4 In this sense, the introduction of trifluoromethyltrimethylsilane (TMSCF3, Ruppert–Prakash reagent)5 allowed productive trifluoromethylations under nucleophilic regimes by exploiting the stabilizing effect imparted by the silicon atom. Analogous nucleophilic difluoromethylations6 and monofluoromethylations7 remained somehow obscured and thus, underdeveloped until recently because of the high tendency of MCHF2 and MCH2F carbanions to undergo α-elimination. On the other hand, the introduction of difluoromethyltrimethylsilane (TMSCHF2), a commercially available and experimentally convenient CHF2-donor source (bp 65 °C)8 boosted the flourishing of synthetic protocols for the introduction of this group9 featuring some unique properties – H-bond donor, weakly acidic, lipophilic isostere of OH and SH motifs – which make it highly valuable inter alia in drug design.10 Compared to the Ruppert–Prakash reagent, the reactivity of TMSCHF2 is tamed11 and its proper activation under Lewis basic conditions is essential, as demonstrated by Hu in 2011 in the course of difluoromethylations of ketones and imines,12 and later extended also to other sp2-hybridized carbon electrophiles by our group.13 Collectively, these precedents showcase that replacing a putative ionic (e.g. Li) M-CHF2 bond with a covalent one (e.g. Si) represents the conditio sine qua non for accessing bench stable difluoromethylating agents. With this rationale in mind, we wondered if a unified strategy enabling the release of the nucleophilic CHF2 moiety from a competent donor to a recipient heteroatom-centered electrophile – [Z]-LG, Z = heteroatom, LG = leaving group – could be designed. Should this concept be experimentally validated, we would establish a smooth access to versatile Z-CHF2 agents not relying on more complex routes such as the Prakash-Olah modification14 of the Cullen CF2 carbene insertion into the Sn-H bond of a trialkyltin hydride.15 Herein, we present the feasibility of this rationale through an alkoxide mediated activation of TMSCHF2: we anticipate that the protocol – working like a CHF2 shuttle – enables the preparation in high chemical yields of α,α-difluoromethyl-derivatives of diverse heteroatoms.
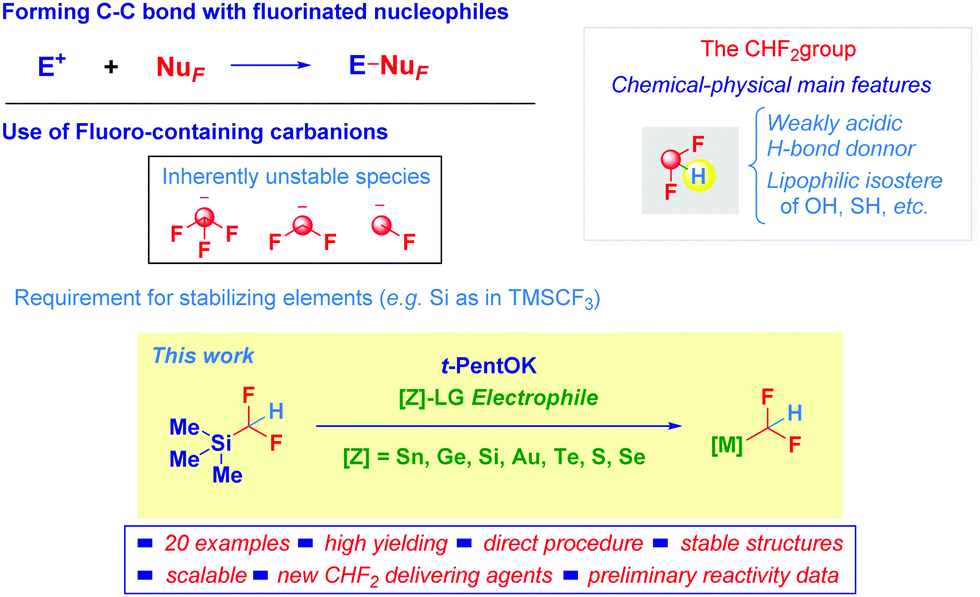 | ||
| Scheme 1 General context of the presented work. | ||
We selected the commercially available chloro-stannatrane 1 as the model substrate for evaluating the strategy proposed (Table 1). This choice was motivated by the following reasons: (a) the stannatrane backbone – introduced in synthesis by Vedejs16 – due to the constitutive apical nitrogen atom which enlarges the Sn–C bond, manifests a higher tendency to transmetallate and thus, to be engaged in nucleophilic transfer operations;17 (b) as showcased in illuminating works by Biscoe,18 stannatranes are particularly suited for coupling (enantioenriched) secondary systems; (c) the expected difluoromethyl analogue 2 is, to the best of our knowledge, an unknown reagent, potentially useful in fluorination chemistry, thus substituting inherently less reactive “dummy”-based species (e.g. R3SnCHF2).19
|
|
||||
|---|---|---|---|---|
| Entry | Activator (equiv.) | TMSCHF2 (equiv.) | Solvent/Temp. (°C) | Yield of 2 (%)a |
| a Yields refer to isolated and purified compound on a model reaction run at 0.68 mmol scale. b 1H-NMR and GCMS analyses of the reaction crude indicate the presence of a stannatrane-O-t-Pent adduct whose purification was unfortunately not effective. c Reaction carried out under non Barbier-type conditions. Entries 1–3 run for 6 h. Entries 4–10 run for 1 h. | ||||
| 1 | CsF (1.8) | 2.0 | Toluene/90 | — |
| 2 | CsF (1.8) | 2.0 | DMF/90 | — |
| 3 | TBAT (1.8) | 2.0 | DMF/90 | — |
| 4 | t-PentOK (1.8) | 2.0 | THF/-50 | 79 |
| 5 | t-PentOK (1.8) | 2.0 | THF/-20 | 87 |
| 6 | t-PentOK (1.8) | 2.0 | THF/0 | 94 |
| 7 | t-PentOK (1.4) | 1.5 | THF/0 | 91 |
| 8 | t-PentOK (1.2) | 1.1 | THF/0 | 83 |
| 9b | t-PentOK (1.5) | 1.5 | THF/0 | 85 |
| 10c | t-PentOK (1.4) | 1.5 | THF/0 | — |

Activating the pronucleophile with CsF (in toluene or DMF) or with TBAT (tetrabutylammonium difluorodiphenylsilicate) was not effective and, the starting chloro-stannatrane 1 was fully recovered (entries 1–3). The adoption of a Lewis base activation protocol with a commercially available solution of potassium tert-pentoxide (amylate) in toluene enabled a clean transformation in THF at −50 °C, thus giving 2 in 79% isolated yield (entry 4). Increasing the temperature – coeteris paribus – to −20 °C and 0 °C, respectively, was beneficial (entries 5–6). The stoichiometric ratio between TMSCHF2 and t-PentOK could be dwindled to 1.5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1.4 without significantly affecting the transformation efficiency (entry 7), whereas a further decrease was detrimental (entry 8). Some additional aspects merit mention: (a) a slight excess (0.1 equiv.) of the pronucleophile compared to the alkoxide was essential for the complete genesis of the difluoromethyl carbanion-like species and thus, for suppressing the (non isolable) stannatrane nucleophilic substitution adduct [Sn–O(t-Pent), entry 9]; (b) using Barbier-type conditions was crucial for observing reactivity, thus remarking the limited chemical integrity of this carbanion (entry 10).
:1.4 without significantly affecting the transformation efficiency (entry 7), whereas a further decrease was detrimental (entry 8). Some additional aspects merit mention: (a) a slight excess (0.1 equiv.) of the pronucleophile compared to the alkoxide was essential for the complete genesis of the difluoromethyl carbanion-like species and thus, for suppressing the (non isolable) stannatrane nucleophilic substitution adduct [Sn–O(t-Pent), entry 9]; (b) using Barbier-type conditions was crucial for observing reactivity, thus remarking the limited chemical integrity of this carbanion (entry 10).
With the optimal conditions for the direct homologative transfer of the CHF2 unit to a halostannane-type derivative,20 we then investigated the scope of the reaction (Scheme 2). Pleasingly, tricyclohexyl- and tri(n-butyl)-stannanes smoothly underwent the transformation, furnishing analogues 3 and 4 in comparable high yield. Switching to aromatic substituents (5) on tin did not affect the effectiveness. Previously undisclosed difluoromethyl derivatives of organogermanium compounds could also be prepared under our conditions in the case of both trialkyl-(6) and triphenyl-(7) systems. This is particularly intriguing since organogermaniums recently emerged as more sustainable and attractive alternatives to the more common organotin compounds.21 Notably, dichlorodiphenylgermanium was a competent electrophile for the double functionalization, conducting to the bis-(difluoromethyl) derivative 8 in a very good 83% isolated yield. The difluoromethyl fragment released by the silicon atom of TMSCHF2 could be efficiently transferred to a different silicon center by reacting with a halo-silane, thus conducting to the unprecedented difluoromethylsilanes 9 (t-butyldiphenyl, 78%) and 10 (triphenyl, 81%). The combined electronic and steric factors imparted by these substituents may be advantageously employed for modulating the reactivity of difluoromethylsilane (vide infra). Furthermore, the NHC–Au(I)–Cl complex could be engaged in the transformation, giving the corresponding –CHF2 adduct 11 in high yield. The scalability of the transformation (15 mmol) was deducted by high-yielding processes for compounds 2 (90%) and 10 (85%).
 | ||
| Scheme 2 Difluoromethyl-group transfer under the nucleophilic regime from TMSCH2 to different heteroatom-based electrophiles. | ||
The formal nucleophilic substitution process was not only achieved on heteroatom-halide functionalities but, was also effective in the case of symmetrical RZ-ZR moieties acting as convenient starting materials. An organotellurium analogue smoothly underwent the reaction, giving (12) in a very good 90% isolated yield. Organosulfur compounds acted as competent substrates for the transformation regardless of the different electronic behaviour displayed by the substituent on the aromatic ring, as evidenced by reactions involving the p-methoxy-(13) and the 2,4,5-trichloro-(14) systems. Notably, engaging a highly sterically hindered material (2,4,6-tri-i-propyl, 15) aromatic disulfide further validated the protocol. Moreover, an excellent chemoselective profile was deducted in the presence of a nitro moiety which remained unaffected, thus furnishing exclusively the difluoromethyl sulfide (16). We further extended the technique to diselenides, it being applicable to both aromatic (17–20) and alkyl analogues (21). The proposed strategy favourably compares with reported protocols, as for example the use of the non-commercially available PhSeCN with the same TMSCHF222 or, the use of the gaseous species chlorodifluoromethane.23
With the aim to gain insights into structural features of difluoromethyl-tin analogues 2 and 5, their crystallographic X-ray analysis revealed some important aspects (Fig. 1). In the case of stannatrane, the Sn1–C1 bond has a length of 2.233 Å, significantly longer compared to classical organotins (R3SnR1).24 This element is in agreement with the reasons accounting for the chemical profile of the stannatrane backbone: the enlarged Sn–C bond makes it more labile and thus, confers a high reactivity. In fact, the analogous bond Sn1–C1 in the triphenyltin analogue 5 is 2.197 Å. Moreover, the Sn1–N1 distance in the stannatrane is 2.478 Å, whereas the two carbon-fluorine bonds in both structures are comparable (1.313 Å and 1.317 Å in stannatrane 2 and, 1.225 Å and 1.338 Å in triphenyltin- 5). The angle C1–Sn1–C2 (102.66°) matches with the analogous C1–Sn1–C5 (103.77°) and C1–Sn1–C8 (102.27°) in the stannatrane, which also shows a characteristic planar orientation N1–Sn1–C6 (178.61°). The absence of the rigidifying backbone in 5 is evident from the values of the angles C1–Sn1–C8 (107.36°), C1–Sn1–C14 (110.30°) and C1–Sn1–C2 (103.13°).
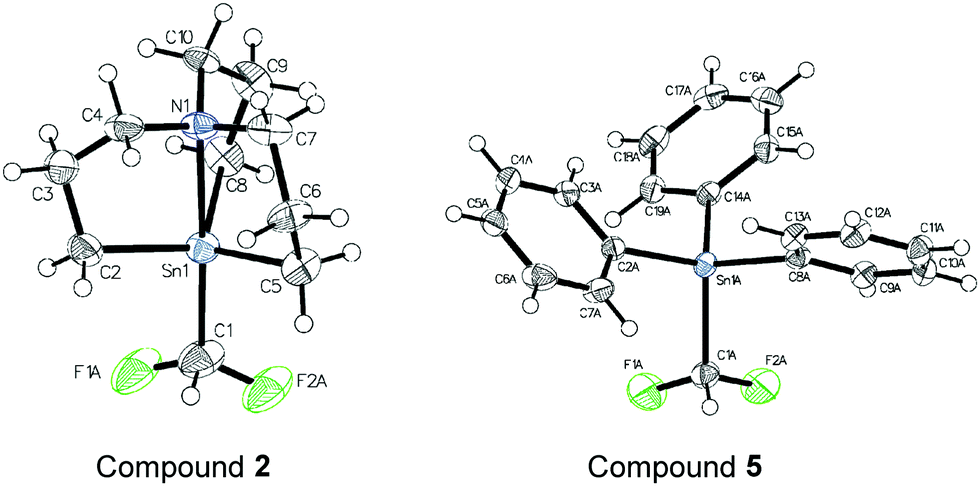 | ||
| Fig. 1 X-ray structures of selected difluoromethyltin derivatives (2 CCDC 2150362 and, 5 CCDC 2150363). | ||
The synthetic potential of selected prepared compounds was then evaluated (Scheme 3). Difluoromethyl stannatrane 2 acted as a versatile coupling agent in two recently developed protocols by Biscoe,18a,d namely: (a) the Pd-catalyzed cross-coupling with bromobenzene furnishing 22 in 76% yield and, (b) the Pd-catalyzed acylation with an acyl chloride 23 which resulted in the clean formation of difluoromethylketone 24 in 83% yield (path a). As an additional proof of the enhanced reactivity conferred by the stannatrane backbone, it is worth noting that Sn-CHF2 analogues 3, 4 and 5 did not promote at any extent both transformations. Intrigued by the solid physical state of the triphenylsilane derivative 10, upon the usual activation with potassium tert-pentoxide and reaction with α,α,α-trifluoroacetophenone 25, we were delighted to observe the preparation of the gem-difluoromethyl-trifluoromethyl carbinol 26 in 87% yield (path b).25 Although reactive, the process carried out with the tert-butyldiphenyl analogue 9, gave 26 in 54% yield, probably as a consequence of the increased steric hindrance on the Si-atom. The same activated form of the triphenylsilane derivative 10 accomplished also a double nucleophilic attack on the azomethinic carbon of N-phenylbenzimidoyl chloride 27en route to bis(difluoromethyl) amine 28 (81% yield). Collectively, these experiments indicate derivative 10 as a valuable difluoromethylating agent form whose solid state – although not suitable for X-ray analysis – may have advantages on the liquid TMSCHF2.
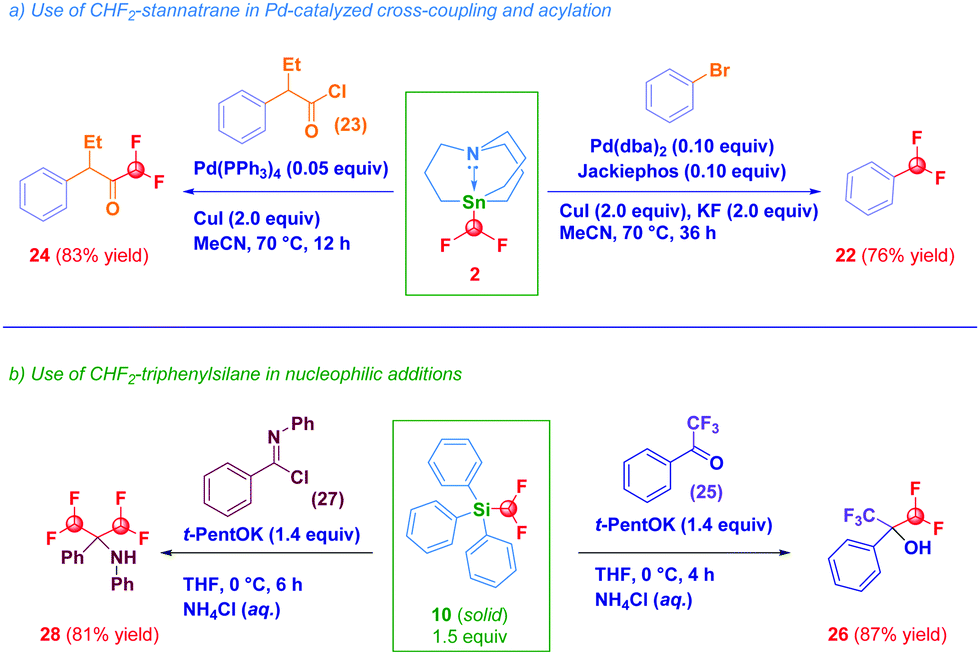 | ||
| Scheme 3 Synthetic uses of CHF2-stannatrane and CHF2-triphenylsilane. | ||
In summary, we reported the direct nucleophilic transfer of a difluoromethyl unit to a series of heteroatom-centered electrophiles (Sn, Ge, Si, Au, Se, S, Te) for forging bench stable analogues. The procedure is levered on the Lewis base mediated activation (potassium tert-pentoxide) of the commercially available and experimentally convenient TMSCHF2. Not only chlorinated starting materials could be employed, but also chalcogenides of general structure RZ-ZR, thus giving straightforward access to the title compounds through a flexible and intuitive logic. Among the prepared motifs, the following deserve a particular mention: (i) the stannatrane analogue for which structural aspects deducted by the X-ray crystallographic analysis support a unique reactivity in Pd-catalyzed cross-coupling or acylation processes and, (ii) the triphenylsilyl-derivative, which can be regarded as a valuable alternative to the starting TMSCHF2 for the delivery of the CHF2 unit to electrophilic linchpins (ketone and imidoyl chloride).
Dedicated to Professor Helmut Spreitzer in on the occasion of his retirement.
The authors thank the University of Vienna, the University of Torino and FWF (project P33130) for financial support.
Conflicts of interest
There are no conflicts to declare.Notes and references
- For authoritative references: (a) J. Wang, M. Sánchez-Roselló, J. L. Aceña, C. del Pozo, A. E. Sorochinsky, S. Fustero, V. A. Soloshonok and H. Liu, Chem. Rev., 2014, 114, 2432 CrossRef CAS PubMed; (b) Y. Zhou, J. Wang, Z. Gu, S. Wang, W. Zhu, J. L. Aceña, V. A. Soloshonok, K. Izawa and H. Liu, Chem. Rev., 2016, 116, 422 CrossRef CAS PubMed; (c) T. Liang, C. N. Neumann and T. Ritter, Angew. Chem., Int. Ed., 2013, 52, 8214 CrossRef CAS PubMed; (d) C. N. Neumann and T. Ritter, Angew. Chem., Int. Ed., 2015, 54, 3216 CrossRef CAS PubMed; (e) D. O'Hagan, Chem. Soc. Rev., 2008, 37, 308 RSC; (f) R. Szpera, D. F. J. Moseley, L. B. Smith, A. J. Sterling and V. Gouverneur, Angew. Chem., Int. Ed., 2019, 58, 14824 CrossRef CAS PubMed.
- For comprehensive discussions, see: (a) J. Hu, W. Zhang and F. Wang, Chem. Commun., 2009, 7465 RSC; (b) R. Britton, V. Gouverneur, J.-H. Lin, M. Meanwell, C. Ni, G. Pupo, J.-C. Xiao and J. Hu, Nat. Rev. Methods Primers, 2021, 1, 47 CrossRef CAS; (c) Emerging Fluorinated Motifs: Synthesis, Properties, and Applications, ed., D. Cahard and J.-A. Ma, Wiley-VCH, Weinheim, 2020 Search PubMed; (d) T. Charvillat, P. Bernardelli, M. Daumas, X. Pannecoucke, V. Ferey and T. Besset, Chem. Soc. Rev., 2021, 50, 8178 RSC; (e) E. Carbonnel, T. Poisson, P. Jubault, X. Pannecoucke and T. Besset, Front. Chem., 2019, 7 Search PubMed.
- For the concept of using metalated α-halogenated synthons, see: (a) L. Castoldi, S. Monticelli, R. Senatore, L. Ielo and V. Pace, Chem. Commun., 2018, 54, 6692 RSC; (b) L. Ielo, V. Pillari, M. Miele, D. Castiglione and V. Pace, Synlett, 2021, 551 CrossRef CAS For recent examples from our group, see: ; (c) R. Senatore, M. Malik, T. Langer, W. Holzer and V. Pace, Angew. Chem., Int. Ed., 2021, 60, 24854 CrossRef CAS PubMed; (d) L. Ielo, L. Castoldi, S. Touqeer, J. Lombino, A. Roller, C. Prandi, W. Holzer and V. Pace, Angew. Chem., Int. Ed., 2020, 59, 20852 CrossRef CAS PubMed; (e) L. Ielo, S. Touqeer, A. Roller, T. Langer, W. Holzer and V. Pace, Angew. Chem., Int. Ed., 2019, 58, 2479 CrossRef CAS PubMed; (f) V. Pace, L. Castoldi, E. Mazzeo, M. Rui, T. Langer and W. Holzer, Angew. Chem., Int. Ed., 2017, 56, 12677 CrossRef CAS PubMed.
- (a) A. D. Dilman and V. V. Levin, Acc. Chem. Res., 2018, 51, 1272 CrossRef CAS PubMed; (b) G. K. S. Prakash and J. Hu, Acc. Chem. Res., 2007, 40, 921 CrossRef CAS PubMed.
- X. Liu, C. Xu, M. Wang and Q. Liu, Chem. Rev., 2015, 115, 683 CrossRef CAS PubMed.
- J. B. I. Sap, C. F. Meyer, N. J. W. Straathof, N. Iwumene, C. W. am Ende, A. A. Trabanco and V. Gouverneur, Chem. Soc. Rev., 2021, 50, 8214 RSC.
- Probably monofluoromethylating agents are the most elusive carbanion-like species, see: (a) G. Parisi, M. Colella, S. Monticelli, G. Romanazzi, W. Holzer, T. Langer, L. Degennaro, V. Pace and R. Luisi, J. Am. Chem. Soc., 2017, 139, 13648 CrossRef CAS PubMed; (b) S. Monticelli, M. Colella, V. Pillari, A. Tota, T. Langer, W. Holzer, L. Degennaro, R. Luisi and V. Pace, Org. Lett., 2019, 21, 584 CrossRef CAS PubMed For a review, see; (c) M. Reichel and K. Karaghiosoff, Angew. Chem., Int. Ed., 2020, 59, 12268 CrossRef CAS PubMed.
- M. Miele and V. Pace, Aust. J. Chem., 2021, 74, 623 CrossRef CAS.
- For comprehensive reviews, see: (a) D. E. Yerien, S. Barata-Vallejo and A. Postigo, Chem. – Eur. J., 2017, 23, 14676 CrossRef CAS PubMed; (b) N. Levi, D. Amir, E. Gershonov and Y. Zafrani, Synthesis, 2019, 4549 CAS . For ground-breaking works on CHF2 chemistry, see: ; (c) P. S. Fier and J. F. Hartwig, J. Am. Chem. Soc., 2012, 134, 5524 CrossRef CAS PubMed; (d) L. Xu and D. A. Vicic, J. Am. Chem. Soc., 2016, 138, 2536 CrossRef CAS PubMed; (e) M. Bos, W.-S. Huang, T. Poisson, X. Pannecoucke, A. B. Charette and P. Jubault, Angew. Chem., Int. Ed., 2017, 56, 13319 CrossRef CAS PubMed.
- (a) C. D. Sessler, M. Rahm, S. Becker, J. M. Goldberg, F. Wang and S. J. Lippard, J. Am. Chem. Soc., 2017, 139, 9325 CrossRef CAS PubMed; (b) Y. Zafrani, G. Sod-Moriah, D. Yeffet, A. Berliner, D. Amir, D. Marciano, S. Elias, S. Katalan, N. Ashkenazi, M. Madmon, E. Gershonov and S. Saphier, J. Med. Chem., 2019, 62, 5628 CrossRef CAS PubMed.
- T. Hagiwara and T. Fuchikami, Synlett, 1995, 717 CrossRef CAS.
- Y. Zhao, W. Huang, J. Zheng and J. Hu, Org. Lett., 2011, 13, 5342 CrossRef CAS PubMed.
- (a) M. Miele, A. Citarella, N. Micale, W. Holzer and V. Pace, Org. Lett., 2019, 21, 8261 CrossRef CAS PubMed; (b) A. Citarella, D. Gentile, A. Rescifina, A. Piperno, B. Mognetti, G. Gribaudo, M. T. Sciortino, W. Holzer, V. Pace and N. Micale, Int. J. Mol. Sci., 2021, 22, 1398 CrossRef CAS PubMed; (c) M. Miele, R. D’Orsi, V. Sridharan, W. Holzer and V. Pace, Chem. Commun., 2019, 55, 12960 RSC; (d) M. Miele, A. Citarella, T. Langer, E. Urban, M. Zehl, W. Holzer, L. Ielo and V. Pace, Org. Lett., 2020, 22, 7629 CrossRef CAS PubMed.
- G. K. S. Prakash, S. K. Ganesh, J.-P. Jones, A. Kulkarni, K. Masood, J. K. Swabeck and G. A. Olah, Angew. Chem., Int. Ed., 2012, 51, 12090 CrossRef CAS PubMed.
- W. R. Cullen, J. R. Sams and M. C. Waldman, Inorg. Chem., 1970, 9, 1682 CrossRef CAS.
- E. Vedejs, A. R. Haight and W. O. Moss, J. Am. Chem. Soc., 1992, 114, 6556 CrossRef CAS.
- (a) N. Srivastav, R. Singh and V. Kaur, RSC Adv., 2015, 5, 62202 RSC; (b) J. G. Verkade, Acc. Chem. Res., 1993, 26, 483 CrossRef CAS; (c) E. Fillion and N. J. Taylor, J. Am. Chem. Soc., 2003, 125, 12700 CrossRef CAS PubMed; (d) A. Kavoosi and E. Fillion, Angew. Chem., Int. Ed., 2015, 54, 5488 CrossRef CAS PubMed.
- (a) C.-Y. Wang, G. Ralph, J. Derosa and M. R. Biscoe, Angew. Chem., Int. Ed., 2017, 56, 856 CrossRef CAS PubMed; (b) C.-Y. Wang, J. Derosa and M. R. Biscoe, Chem. Sci., 2015, 6, 5105 RSC; (c) X. Ma, B. Murray and M. R. Biscoe, Nat. Rev. Chem., 2020, 4, 584 CrossRef CAS PubMed; (d) L. Li, C.-Y. Wang, R. Huang and M. R. Biscoe, Nat. Chem., 2013, 5, 607 CrossRef CAS PubMed.
- (a) C. Cordovilla, C. Bartolomé, J. M. Martínez-Ilarduya and P. Espinet, ACS Catal., 2015, 5, 3040 CrossRef CAS; (b) S. Touqeer, L. Castoldi, T. Langer, W. Holzer and V. Pace, Chem. Commun., 2018, 54, 10112 RSC.
- E. Le Grognec, J.-M. Chrétien, F. Zammattio and J.-P. Quintard, Chem. Rev., 2015, 115, 10207 CrossRef CAS PubMed.
- M.-Y. Xu, W.-T. Jiang, Y. Li, Q.-H. Xu, Q.-L. Zhou, S. Yang and B. Xiao, J. Am. Chem. Soc., 2019, 141, 7582 CrossRef CAS PubMed.
- T. Dong, J. Nie and C.-P. Zhang, Tetrahedron, 2018, 74, 5642 CrossRef CAS.
- H. Suzuki, M. Yoshinaga, K. Takaoka and Y. Hiroi, Synthesis, 1985, 497 CrossRef CAS.
- F. H. Allen, O. Kennard, D. G. Watson, L. Brammer, A. G. Orpen and R. Taylor, J. Chem. Soc., Perkin Trans. 2, 1987, S1 RSC.
- S. Meyer, J. Häfliger and R. Gilmour, Chem. Sci., 2021, 12, 10686 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2150362–2150363. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2cc00886f |
| This journal is © The Royal Society of Chemistry 2022 |