
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Order, disorder, and metalation of tetraphenylporphyrin (2H-TPP) on Au(111)†
Matthew
Edmondson
 a,
Eleanor S.
Frampton
ab,
Chris J.
Judd
a,
Neil R.
Champness
c,
Robert G.
Jones
b and
Alex
Saywell
*a
a,
Eleanor S.
Frampton
ab,
Chris J.
Judd
a,
Neil R.
Champness
c,
Robert G.
Jones
b and
Alex
Saywell
*a
aSchool of Physics & Astronomy, The University of Nottingham, Nottingham NG7 2RD, UK. E-mail: Alex.Saywell@nottingham.ac.uk
bSchool of Chemistry, The University of Nottingham, Nottingham NG7 2RD, UK
cSchool of Chemistry, University of Birmingham, Edgbaston, Birmingham, B15 2TT, UK
First published on 22nd April 2022
Abstract
A thermally induced order–disorder transition of tetraphenylporphyrin (2H-TPP) on Au(111) is characterised by scanning probe microscopy and X-ray photoelectron spectroscopy-based techniques. We observed that a transition from an ordered close-packed phase to a disordered diffuse phase is correlated with an on-surface cyclodehydrogenation reaction, and that additional heating of this diffuse phase gives rise to a single distinct nitrogen environment indicative of the formation of a Au–TPP species.
Porphyrins are π-conjugated macrocycles which may exhibit bespoke chemical functionality via the addition of specific chemical groups at the periphery of the molecule. Incorporation of a metal atom within the macrocycle cavity provides further functionality and access to applications based upon catalysis1,2 and sensing.3 Porphyrins, and associated derivatives, have been extensively studied by a range of surface-sensitive techniques,4 including scanning probe microscopies.5 Of particular interest is the metalation of porphyrins, with various strategies being employed, including: solution phase synthesis,6 metal-uptake via co-deposition of metal and porphyrin species,7,8 and self-metalation involving sequestration of metal atoms from a substrate.9–12
The archetypal example of thermally activated self-metalation is the interaction of 2H-tetraphenylporphyrin (2H-TPP) with Cu(111).10,13 During self-metalation of 2H-TPP, the two distinct nitrogen environments (iminic, ![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N–, and pyrrolic, –NH–) are replaced by a single environment with the loss of hydrogen atoms and all nitrogen atoms interacting with a central metal atom.13 Other thermally activated reactions may occur which change the structure of the macrocycle, such as cyclodehydrogenation of the ring structure (removal of hydrogen and formation of C–C bonds between the porphine and phenyl groups which flattens the porphyrin conformation).14,15
N–, and pyrrolic, –NH–) are replaced by a single environment with the loss of hydrogen atoms and all nitrogen atoms interacting with a central metal atom.13 Other thermally activated reactions may occur which change the structure of the macrocycle, such as cyclodehydrogenation of the ring structure (removal of hydrogen and formation of C–C bonds between the porphine and phenyl groups which flattens the porphyrin conformation).14,15
The self-metalation of 2H-TPP on Au(111) is less well studied, partly due to the perception that gold is a relatively inert substrate. However, the uptake of gold into porphyrin macrocycles, to form a Au-TPP species, has recently been reported,9 and novel methods of gold porphyrin synthesis are relevant due to a possible role as a functional species within anticancer complexes.16
Here we report an order to disorder transition of 2H-TPP assemblies on Au(111). Conversion from a close-packed phase to a diffuse phase is initiated via annealing, and we discuss the transition mechanism in-light of a detailed study incorporating complementary techniques; X-ray photoelectron spectroscopy (XPS), near edge X-ray absorption fine structure (NEXAFS), and scanning tunnelling microscopy (STM). This approach allows changes in chemical environments and molecule/surface interactions to be determined. We identify that the order/disorder transition is driven by an on-surface ring-closing reaction, and observe that further annealing results in formation of a single nitrogen chemical environment due to dehydrogenation and subsequent metalation with gold, in agreement with our NEXAFS data.
A close-packed monolayer of 2H-TPP was formed via sublimation onto a Au(111) surface held under ultra-high vacuum (UHV) conditions with the resulting molecular assemblies characterised via STM (see ESI† for full experimental method). Fig. 1a shows an STM topograph acquired at ∼ 80 K where a variety of packing motifs were observed (see ESI†), however, the majority of close-packed structures are consistent with previously reported observations.15,17,18 Annealing to 580 ± 50 K resulted in the loss of the close-packed phase and the formation of a ‘diffuse phase’, shown in Fig. 1b (imaged at 6.5 K). The overall surface coverage of TPP has reduced following annealing (from 0.51 ± 0.05 to 0.43 ± 0.04 molecules per nm2). A similar transition is observed for sub-molecular coverages of TPP (compare Fig. 1c and d), indicating that the observed change is not simply a function of coverage. For sub-monolayer coverages, small close-packed islands (consisting of 2–9 TPP molecules) are observed to form exclusively within the face centred cubic (FCC) regions of the herringbone reconstruction. Upon annealing to 580 ± 50 K, isolated single molecules are again found to be present within the FCC regions of the surface, but the formation of islands is not observed. This implies a reduction in the strength of the lateral interaction between molecules which we attribute to a loss of C−H→π interactions19 due to the ring-closing reaction. It has also been reported that free-base porphine has a repulsive long-range interaction (attributed to charge transfer into the molecule);20 this mechanism would also lead to diffuse 2H-TPP structures in concert with the reduction in molecule–molecule cohesion.
 | ||
| Fig. 1 LEED and STM images showing the close-packed and diffuse phases of 2H-TPP on Au(111). (a) monolayer close-packed phase (Iset = 50 pA, Vbias = 0.8 V, 80 K) with inset of 2H-TPP chemical structure, (b) diffuse phase, formed by annealing a monolayer of 2H-TPP to 580 ± 50 K (Iset = 5 pA, Vbias = 1.3 V, 6.5 K), (c) sub-monolayer close-packed phase (Iset = 50 pA, Vbias = −1.3 V, 6.5 K) and (d) annealed sub-monolayer resulting in the diffuse phase (Iset = 25 pA, Vbias = 1.3 V, 6.5 K). (e and f) LEED patterns acquired at 297 K for 2H-TPP on Au(111), (e) monolayer close-packed phase (LEED energy 15.5 eV) and (f) an annealed monolayer giving rise to the diffuse phase (LEED energy 6.5 eV). | ||
Additional information on the transition between close-packed and diffuse phases is obtained via low energy electron diffraction (LEED) measurements. LEED data for the close-packed TPP monolayer (Fig. 1e) indicates an ordered phase with a non-commensurate lattice, in agreement with STM measurements (see ESI†). Conversely, LEED data for the diffuse phase (Fig. 1f) shows a broad ring indicative of a lack of long-range order.
We interpret the order–disorder transition as an on-surface process resulting in a change to the 2H-TPP molecule–molecule interaction. On-surface cyclodehydrogenation of 2H-TPP, resulting in ‘ring-closing’ between the phenyl groups and the porphine ring, has previously been reported.14,15 Our STM analysis of 2H-TPP on Au(111) indicates that annealed TPP, in the diffuse phase, has a different structure (see Fig. 1b and Fig. S6, ESI†) to that in the close-packed phase; similar to that reported for a fluorinated variant of 2H-TPP.9
A combination of XPS, LEED, and NEXAFS measurements were performed at Diamond Light Source (Oxfordshire, UK – experimental details in ESI†) to further investigate the on-surface processes. A monolayer coverage of 2H-TPP was prepared on a Au(111) single crystal and the presence of a close-packed structure at room temperature confirmed via LEED (see Fig. 1e). Heating the sample to 440 ± 50 K produced no observable effect on the LEED pattern (indicating stability of the close-packed structure). XPS data showing the C1s environments (shown in Fig. 2a and spectra in Fig. 2b.) were assigned using a similar methodology to Nardi et al.21 The C1s region (shown in Fig. 2b), is fitted using a combination of four environments assigned to: the 4 phenyl rings (CPh), at 284.1 eV binding energy (BE), the carbons at the meso (Cm) and CBα positions, at 284.7 eV BE,CAα carbons, at 285.5 eV BE, and CAβ and CBβ at 283.9 eV BE. A shakeup feature is visible at ∼ 287 eV BE, but is negligible within our analysis. These binding energies were referenced to the Fermi level of the Au surface. The absolute BEs observed, unsurprisingly, differ from those reported by Nardi et al. for a film of 2H-TPP on SiO2/Si(100), however, there is excellent agreement with respect to the relative shifts between the assigned carbon environments.21 Similar to the behaviour observed in the LEED analysis, there was no change in the C1s peak following heating the sample to 440 ± 50 K.
 | ||
| Fig. 2 (a) Assignment of carbon environments within the 2H-TPP molecule based on work by Nardi et al.21 (b) C1s XP spectra for the close-packed phase of 2H-TPP on Au(111), based upon the assignments in (a), showing a peak structure consistent with previous reports.21 | ||
N1s XP spectra obtained for the close-packed TPP structure (shown in Fig. 3a), contains components assigned to iminic groups (N–), at 397.2 eV BE, and pyrrolic (–NH–) groups, at 399.3 eV BE (with the expected ∼ 2 eV shift between the two peaks observed). Additional features at 397.7 eV and 400.0 eV BE are also present. While it is possible that these features relate to multilayers of TPP,13 we observed an increase in the relative intensity of these peaks following annealing to 440 ± 50 K. We therefore assign these features to an additional interaction between the iminic and pyrrolic nitrogens and a gold species. Based upon previous STM and density functional theory (DFT) studies of 2H-TPP on Au(111) we assign the interaction to a gold adatom-2H-TPP species:17 where the co-existence of 2H-TPP and adatom-2H-TPP was confirmed using scanning tunnelling spectroscopy (STS). Here, our STS characterisation of the surface allows the identification, and demonstrates the co-existence, of the two porphyrin species (see Fig. S8, ESI†). Metal adatoms are known to diffuse freely on metal surfaces, including Au(111), with diffusion away from step-edges a likely source of Au species.22,23 In common with previous studies we find no evidence for disruption of the Au(111) herringbone reconstruction.9 Additionally, we note that the ratio of the 2H-TPP to adatom-2H-TPP species (as identified within the N1s XPS data) varies with sample preparation and upon annealing; in line with the expectation that the availability of Au adatoms is related to the surface temperature.
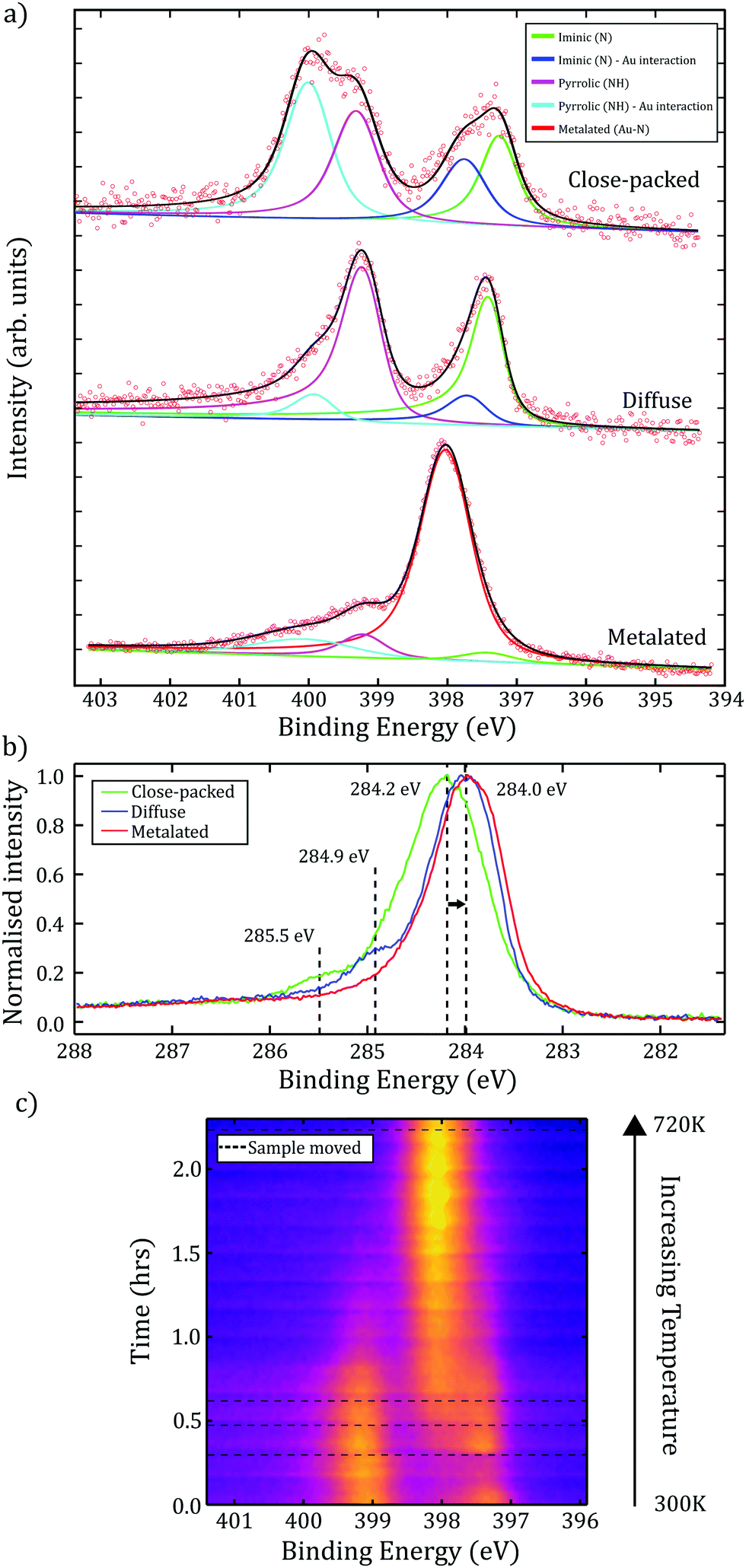 | ||
| Fig. 3 (a) XP spectra for the N1s environment for each phase of TPP (close-packed/diffuse/metalated). Within close-packed and diffuse spectra, iminic and pyrrolic groups are present; additional peaks at higher BEs due to interaction with Au adatoms. Annealing the diffuse phase produced a single nitrogen feature, indicative of a deprotonated environment within a metalated Au–TPP species. (b) C1s XP spectra showing a shift to lower BE and loss of high BE features following annealing. (c) Time-resolved N1s XPS starting with the diffuse preparation at 0.0 hours. Sample heated to 720 ± 50 K over 2 hours (non-linear temperature change), resulting in a single metalated N1s peak. Dotted lines correspond to sample movement to reduce the effect of beam-damage. | ||
The diffuse phase was obtained by annealing the surface whilst performing in situ MCP-LEED, with the transition estimated to occur at 540 ± 50 K. XP spectra of the C1s region show evidence for two important changes (see Fig. 3b); a shift of the major component to a lower binding energy (by ≈ 0.2 eV), indicating a change in the environment of the phenyl rings, and the loss of a feature at 285.5 eV (assigned to CAα). This is consistent with a 2H-TPP ring-closing reaction,14 and agrees with our STM observations of ‘distorted’ 2H-TPP species (see ESI†) following annealing. Related studies discussing fluorinated TPP species on Au(111),9 2H-TPP on Au(111)15 and 2H-TPP on Ag(111),14 report that phenyl rings are likely to react with the iminic part of the porphine ring (where as the loss of the CAα feature suggests a reaction via the pyrrolic part of the ring). Our study is consistent with a ring-closing reaction, and we propose that this is the on-surface process underpinning the observed order–disorder transition. The N1s region of the XP spectra for the diffuse phase (Fig. 3a.) shows a reduction in the intensity of the peaks assigned to adatom-2H-TPP (399.9 eV and 397.7 eV BE), which is in agreement with an interpretation that flattening of the porphyrin species results in a reduction of conformation flexibility (e.g. switching from ‘saddle’ to a more ‘planar’ conformation), and hence reduces the interaction between Au adatoms and the nitrogen species within the porphyrin core.
Previous work characterising 2H-TPP on metallic substrates has shown that self-metalation of the porphyrin may be initiated by annealing.10,13 We explore the effect of annealing via time-resolved XPS measurements to determine changes to the chemical environment of the molecule. The measurements for the N1s region were acquired while annealing to 720 ± 50 K over 2.2 hours. To check the effect of ‘beam-damage’ the sample was periodically moved relative to the incident X-rays (indicated as dashed lines in Fig. 3c). We attribute the peak observed to grow at 398.0 eV to a strong interaction between four dehydrogenated nitrogen atoms within the porphyrin core and a Au species. This single nitrogen environment is indicative of full self-metalation of TPP. The partial metalation of TPP, due to interaction with Au adatoms or the substrate, would be expected to result in a slight increase in BE;24 compatible with the observed shift in BE from 397.7 eV to 398.0 eV. However, although metalated porphyrin species typically have nitrogen BEs in the range of 398.6–398.9 eV, it is known that this may be lowered as a result of interaction with the surface,25 or as an effect of localised charge on the molecule (e.g. Au–TPP has previously been reported to exist in the 3+ oxidation state),26 and as such our results are compatible with the formation of a Au–TPP species.
To further explore the formation of Au–TPP, NEXAFS measurements were performed on each of the three experimentally obtained TPP phases. Changes in the nitrogen K-edge spectra relate to changes in the structure of unoccupied molecular orbitals (σ* and π*) and can provide information on the bonding interactions of the central nitrogen atoms; and therefore provided a ‘fingerprint’ of the metalated TPP species. A direct comparison between our NEXAFS data for the metalated phase and a previously reported characterisation of metalated TPP on Cu(111) (supported by density functional theory, DFT, calculations),25 shows excellent agreement with regards to the presence of three π* resonances at photon energies of 398.4, 400.7, and 401.3 eV (see ESI† for NEXAFS spectra). Comparison between the DFT calculated NEXAFS spectrum for a deprotonated TPP species (i.e. non-metalated) and our data, precludes the possibility that the single nitrogen environment observed in the N1s XP spectra is due to the presence of a deprotonated, but non-metalated, TPP species.
In summary, we have utilised a combination of STM, LEED, XPS and NEXAFS to characterise an order to disorder transition of TPP on Au(111). The surface-sensitive techniques provided information on the chemical and order–disorder changes that occurred during this on-surface process. The observed transition from an ordered close-packed phase to a disordered diffuse phase was found to be correlated with a cyclodehydrogenation ring-closing reaction, which resulted in the ‘flatter’ porphyrin. Analysis of N1s XPS data allowed us to identify two distinct porphyrin species within the close-packed phase, 2H-TPP and adatom-2H-TPP (a 2H-TPP species which forms a strong interaction with a Au adatom); in agreement with previous STM studies of the 2H-TPP Au(111) system. Further annealing of the diffuse phase resulted in formation of a single nitrogen environment which, supported by previous studies and our own NEXAFS measurements, is indicative of the formation of a fully metalated Au-TPP species.
The authors would like to thank Diamond Light Source for the award of beam time (SI23730-1), and Pardeep K. Thakur/David A. Duncan for their assistance. A.S. thanks the Royal Society for support via a University Research Fellowship. NRC gratefully acknowledges the support of the UK Engineering and Physical Sciences Research Council (EP/S002995/2). The experimental data on which this work is based can be found at https://dx.doi.org/10.17639/nott.7167.
Conflicts of interest
The authors declare no conflicts of interest.References
- B. Hulsken, R. Van Hameren, J. W. Gerritsen, T. Khoury, P. Thordarson, M. J. Crossley, A. E. Rowan, R. J. Nolte, J. A. Elemans and S. Speller, Nat. Nanotechnol., 2007, 2, 285–289 CrossRef CAS PubMed.
- C. Costentin, H. Dridi and J. M. Savéant, J. Am. Chem. Soc., 2015, 137, 13535–13544 CrossRef CAS PubMed.
- B. R. Takulapalli, G. M. Laws, P. A. Liddell, J. Andréasson, Z. Erno, D. Gust and T. J. Thornton, J. Am. Chem. Soc., 2008, 130, 2226–2233 CrossRef CAS PubMed.
- J. M. Gottfried, Surf. Sci. Rep., 2015, 70, 259–379 CrossRef CAS.
- W. Auwärter, D. Écija, F. Klappenberger and J. V. Barth, Nat. Chem., 2015, 7, 105–120 CrossRef PubMed.
- E. B. Fleischer and A. Laszlo, Inorg. Nucl. Chem. Lett., 1969, 5, 373–376 CrossRef CAS.
- D. A. Duncan, P. S. Deimel, A. Wiengarten, M. Paszkiewicz, P. Casado Aguilar, R. G. Acres, F. Klappenberger, W. Auwärter, A. P. Seitsonen, J. V. Barth and F. Allegretti, J. Phys. Chem. C, 2019, 123, 31011–31025 CrossRef CAS.
- K. Flechtner, A. Kretschmann, L. R. Bradshaw, M. M. Walz, H. P. Steinrück and J. M. Gottfried, J. Phys. Chem. C, 2007, 111, 5821–5824 CrossRef CAS.
- B. Cirera, B. De La Torre, D. Moreno, M. Ondrácek, R. Zboril, R. Miranda, P. Jelínek and D. Écija, Chem. Mater., 2019, 31, 3248–3256 CrossRef CAS.
- M. Röckert, S. Ditze, M. Stark, J. Xiao, H. P. Steinrück, H. Marbach and O. Lytken, J. Phys. Chem. C, 2014, 118, 1661–1667 CrossRef.
- A. Goldoni, C. A. Pignedoli, G. Di Santo, C. Castellarin-Cudia, E. Magnano, F. Bondino, A. Verdini and D. Passerone, ACS Nano, 2012, 6, 10800–10807 CrossRef CAS PubMed.
- C. M. Doyle, S. A. Krasnikov, N. N. Sergeeva, A. B. Preobrajenski, N. A. Vinogradov, Y. N. Sergeeva, M. O. Senge and A. A. Cafolla, Chem. Commun., 2011, 47, 12134–12136 RSC.
- K. Diller, F. Klappenberger, M. Marschall, K. Hermann, A. Nefedov, C. Wöll and J. V. Barth, J. Chem. Phys., 2012, 136, 014705 CrossRef CAS PubMed.
- A. Wiengarten, J. A. Lloyd, K. Seufert, J. Reichert, W. Auwärter, R. Han, D. A. Duncan, F. Allegretti, S. Fischer, S. C. Oh, Z. Salam, L. Jiang, S. Vijayaraghavan, D. Écija, A. C. Papageorgiou and J. V. Barth, Chem. – Eur. J., 2015, 21, 12285–12290 CrossRef CAS PubMed.
- J. Lu, B. Da, W. Xiong, R. Du, Z. Hao, Z. Ruan, Y. Zhang, S. Sun, L. Gao and J. Cai, Phys. Chem. Chem. Phys., 2021, 23, 11784–11788 RSC.
- I. Toubia, C. Nguyen, S. Diring, L. M. Ali, L. Larue, R. Aoun, C. Frochot, M. Gary-Bobo, M. Kobeissi and F. Odobel, Inorg. Chem., 2019, 58, 12395–12406 CrossRef CAS PubMed.
- J. Mielke, F. Hanke, M. V. Peters, S. Hecht, M. Persson and L. Grill, J. Am. Chem. Soc., 2015, 137, 1844–1849 CrossRef CAS PubMed.
- J. Mielke, J. Martínez-Blanco, M. V. Peters, S. Hecht and L. Grill, Phys. Rev. B, 2016, 94, 35416 CrossRef.
- G. Rojas, X. Chen, C. Bravo, J. H. Kim, J. S. Kim, J. Xiao, P. A. Dowben, Y. Gao, X. C. Zeng, W. Choe and A. Enders, J. Phys. Chem. C, 2010, 114, 9408–9415 CrossRef CAS.
- F. Bischoff, K. Seufert, W. Auwärter, S. Joshi, S. Vijayaraghavan, D. Écija, K. Diller, A. C. Papageorgiou, S. Fischer, F. Allegretti, D. A. Duncan, F. Klappenberger, F. Blobner, R. Han and J. V. Barth, ACS Nano, 2013, 7, 3139–3149 CrossRef CAS PubMed.
- M. Nardi, R. Verucchi, C. Corradi, M. Pola, M. Casarin, A. Vittadini and S. Iannotta, Phys. Chem. Chem. Phys., 2010, 12, 871–880 RSC.
- M. Giesen, Prog. Surf. Sci., 2001, 68, 1–154 CrossRef CAS.
- D. R. Peale and B. H. Cooper, J. Vac. Sci. Technol., A, 1998, 10, 2210 CrossRef.
- J. P. Macquet, M. M. Millard and T. Theophanides, J. Am. Chem. Soc., 1978, 100, 4741–4746 CrossRef CAS.
- K. Diller, A. C. Papageorgiou, F. Klappenberger, F. Allegretti, J. V. Barth and W. Auwärter, Chem. Soc. Rev., 2016, 45, 1629–1656 RSC.
- S. Müllegger, W. Schöfberger, M. Rashidi, L. M. Reith and R. Koch, J. Am. Chem. Soc., 2009, 131, 17740–17741 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: experimental Methods; Additional STM, STS, and XPS details; NEXAFS data. See DOI: https://doi.org/10.1039/d2cc00820c |
| This journal is © The Royal Society of Chemistry 2022 |