
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Supramolecular networks by imine halogen bonding†
Esther
Nieland
 a,
Daniel
Komisarek
b,
Stephan
Hohloch
c,
Klaus
Wurst
c,
Vera
Vasylyeva
b,
Oliver
Weingart
*d and
Bernd M.
Schmidt
*a
a,
Daniel
Komisarek
b,
Stephan
Hohloch
c,
Klaus
Wurst
c,
Vera
Vasylyeva
b,
Oliver
Weingart
*d and
Bernd M.
Schmidt
*a
aInstitut für Organische Chemie und Makromolekulare Chemie, Heinrich-Heine-Universität Düsseldorf, Universitätsstrasse 1, 40225 Düsseldorf, Germany. E-mail: Bernd.Schmidt@hhu.de
bInstitut für Anorganische Chemie und Strukturchemie I, Heinrich-Heine-Universität Düsseldorf, Universitätsstrasse 1, 40225 Düsseldorf, Germany
cInstitut für Allgemeine, Anorganische und Theoretische Chemie, Universität Innsbruck, Innrain 80-82, 6020 Innsbruck, Austria
dInstitut für Theoretische Chemie und Computerchemie, Heinrich-Heine-Universität Düsseldorf, Universitätsstrasse 1, 40225 Düsseldorf, Germany. E-mail: Oliver.Weingart@hhu.de
First published on 30th March 2022
Abstract
Halogen bonding of neutral donors using imine groups of porous organic cage compounds as acceptors leads to the formation of halogen-bonded frameworks. We report the use of two different imine cages, in combination with three electron-poor halogen bond donors. Four resulting solid-state structures elucidated by single-crystal X-ray analysis are presented and analysed for the first time by plane-wave DFT calculations and QTAIM-analyses of the entire unit cells, demonstrating the formation of halogen bonds within the networks. The supramolecular frameworks can be obtained either from solution or mechanochemically by liquid-assisted grinding.
Halogen bonding is a highly directional and tuneable non-covalent interaction.1,2 Anions, π-systems and a variety of lone pair possessing molecules such as, for example, amines,3a dithianes,3b thiocarbonyls,3c isocyanides,3d or phosphines3e,f can act as halogen bond acceptors, leading to numerous diverse and exciting applications of halogen bonding.1c,f,i Chiral fractal architectures with amplified circularly polarized luminescence were generated using halogen bonding,4a while Xing and co-workers recently illustrated the crucial role of halogen bonds in controlling supramolecular chirality in multicomponent systems.4b Supramolecular structures such as macrocycles,5 boxes,6 and capsules7 were assembled using halogen bonding. Employing two resorcin[4]arene-based hemispheres, a supramolecular capsule was formed by halogen bonding.7b,g Rissanen and colleagues used halogen bonding of halonium ions [N⋯I+⋯N] to create capsules of various sizes,1e,7a,c–e including a 4.5 nm cavitand-based hexameric capsule.7c Halonium ion bonding has also recently been used to create a halogen-bonded organic framework.8a Networks based on halogen bonding of neutral halogen bond donors8b–g emit organic room-temperature phosphorescence,8b or were shown to undergo guest-induced structural transformations.8e Self-assembly in the solid state is an important aspect of porous organic cage (POC) compounds.9,10 Because the solid state packing of POCs has a large influence on their properties,9 several groups have developed methods to control the arrangement by covalently linking several POCs to form networks and covalent organic frameworks (COFs) based on POCs.11 Crystallisation of POCs can be directed in selected examples by using specific solvents.12 We herein introduce imine-based halogen bonding for the formation of supramolecular networks.
Unlike many other halogen bond donors, imines have been previously used in catalysis13a–e but no further research regarding their halogen bonding properties or the binding motif itself has been conducted.1h,13f
Using this interaction, no functionalisation of the imine POCs is needed, as all cages formed by dynamic covalent chemistry between aldehydes and amines carry imine groups. The integration of POCs into halogen-bonded frameworks is a technique for facilitating crystallisation and influencing solid-state packing. We exemplarily chose a small Tri2Di3 cage (A) based on isophthalaldehyde and (2,4,6-trimethylbenzene-1,3,5-triyl)trimethanamine, and a larger Tri4Di6 cage (B) synthesised from terephthalaldehyde and (2,4,6-trimethylbenzene-1,3,5-triyl)trimethanamine, as halogen bond acceptors.14,15 Addition of fluorinated halogen bond donors, iodopentafluorobenzene (IF5), 1,4-diiodotetrafluorobenzene (I2F4) and 1,3,5-triiodotrifluorobenzene (I3F3) to these cages resulted in the formation of four different co-crystals with completely different topologies.
To study imine halogen bonding, a model system based on IF5 as a halogen bond donor and N-benzylideneaniline as a halogen bond acceptor (Fig. 1) was chosen and investigated in the gas phase and in solution. Analysis of the DFT-computed density showed interaction energies for the N⋯I halogen bond (2.89 Å, 18.62–29.07 kJ mol−1) and additional H⋯I interactions. In comparison, typical values for halogen bonding lie between 10 kJ mol−1 and 150 kJ mol−1 depending on the chosen donors and acceptors.1h,16 The donor IF5 can also be used to generate a co-crystal with imine cage A in which each cage is connected by imine halogen bonding to one donor (Fig. S3 and S4, Table S4, ESI†).
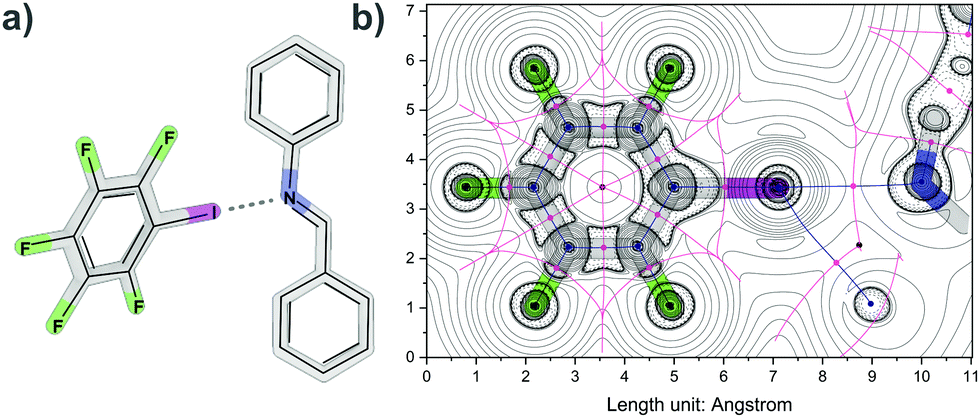 | ||
| Fig. 1 (a) Imine halogen bonding between iodopentafluorobenzene (IF5) and N-benzylideneaniline; (b) corresponding Laplacian of electron density with critical points and bond paths. Bond critical (3, −1) points are depicted in pink, nuclear critical (3, −3) points in blue, ring critical (3, +1) points in black, paths and zero-flux surfaces as blue and pink lines. | ||
After investigating the general halogen bond acceptor properties of imines, we used I2F4 as a donor with a higher count of iodine to build networks based on cage A.
A structure resembling a box is formed by two cages A connected by halogen bonding of four I2F4 molecules (Fig. 2). The four different N⋯I halogen bonds are of similar strength, ranging in length between 2.933(3) and 2.997(5) Å with N⋯I–C angles between 164.6(2) and 171.6(2)° (normalized bond lengths for all XBs, Table S8, ESI†). The unoccupied side of A interacts with neighbouring cages, each forming its own box, via π-stacking and distant π-stacking, building a 2D lattice.
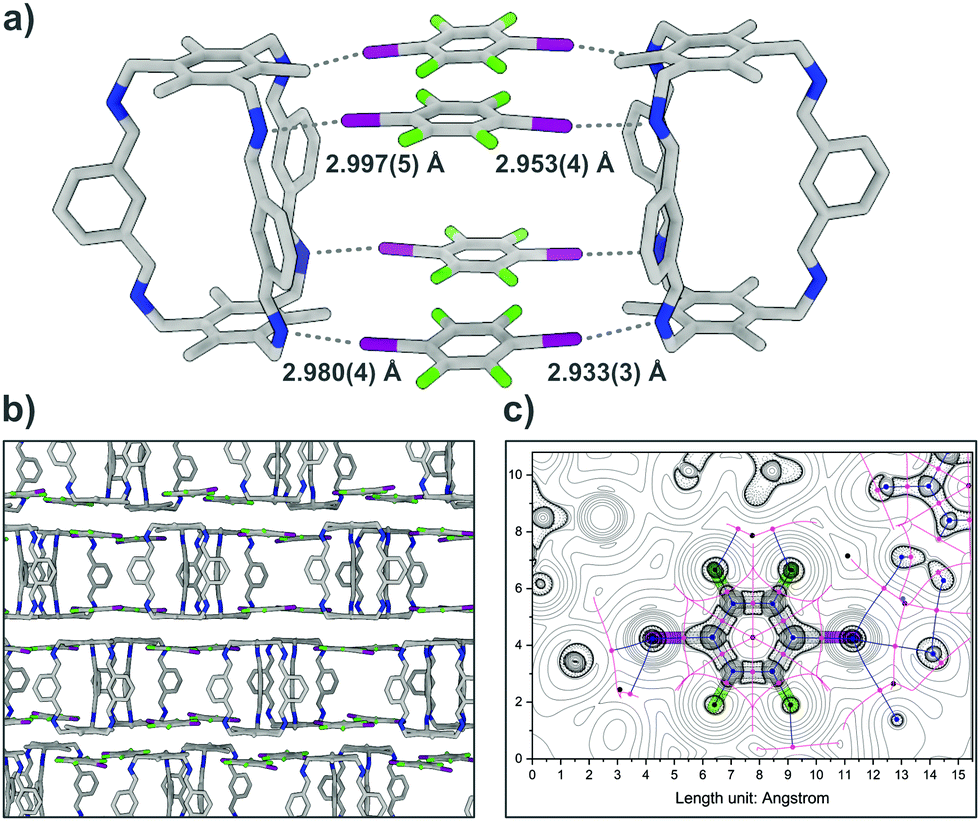 | ||
| Fig. 2 (a) Box-like structure in the single-crystal structure of A⋯2(I2F4), N⋯I halogen bonds are shown as grey dotted lines, (solvent molecules are omitted for clarity); (b) packing of A⋯2(I2F4) along the crystallographic b axis; (c) corresponding Laplacian of electron density with critical points and bond paths. Bond critical (3, −1) points are depicted in pink, nuclear critical (3, −3) points in blue, ring critical (3, +1) points in black, paths and zero-flux surfaces as blue and pink lines. | ||
To confirm that the bonding in A⋯2(I2F4) is indeed intermolecular halogen bonding as suggested by the geometrical parameters, plane-wave DFT-D3 calculations17 were performed, as well as detailed QTAIM-analyses,18 demonstrating the existence of appropriate bond critical points (3, −1) for the N⋯I contacts (Fig. S12, ESI†). The unit cell structure of A⋯2(I2F4) was geometry-optimised, showing marginal shortening of the N⋯I bond to 2.84 and 2.89 Å. The structure was used to calculate the interaction energies for two halogen bonds that lie between 15.64 and 24.43 kJ mol−1 and 17.66 and 27.58 kJ mol−1, respectively. A simplified model system was also created, yielding bond lengths and interaction energies of the same magnitude (Fig. S16 and S17, Table S10, ESI†). In A⋯4(I3F3), each donor site of A is connected via imine halogen bonding to one I3F3, binding to three cages in total, in an infinite 2D network (Fig. 3). The six different N⋯I halogen bonds are of slightly different lengths (distances between 2.967(5) and 3.016(4) Å) and angles (N⋯I–C angles between 163.3(1) and 166.4(1)°). Two additional I3F3 are showing only minor interactions with A. These donors, which act as a “solvent molecule” in the crystal structure, aid in the interconnection of the different layers via halogen bonding with lattice-building and π-stacking with “solvent-like” I3F3.
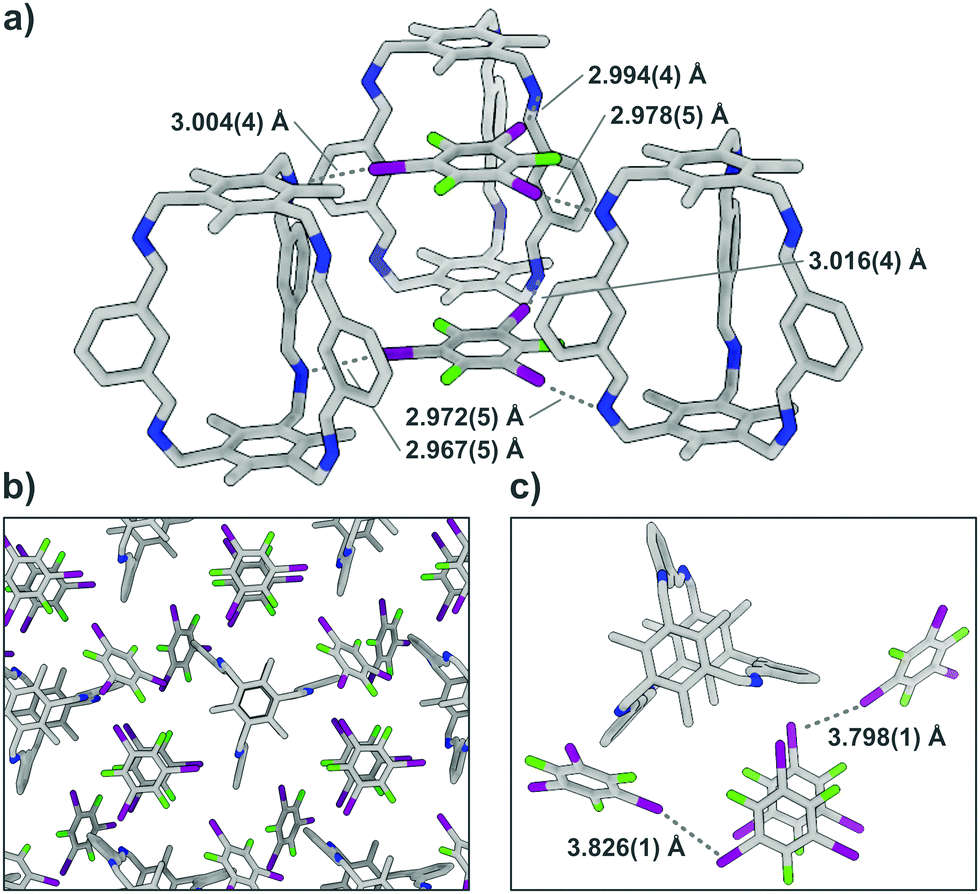 | ||
| Fig. 3 (a) Lattice-building motif in the single-crystal structure of A⋯4(I3F3), N⋯I halogen bonds are shown as grey dotted lines, I3F3 exhibiting no halogen bonds with A are not shown; (b) infinite network built by A⋯4(I3F3) pictured along the crystallographic c axis; (c) cage with lattice-building and “solvent-like” I3F3, additional I⋯I halogen bonds are shown as grey dotted lines. | ||
Theoretical calculations support these findings, as the two lattice-building I3F3 each participate in three intermolecular N⋯I halogen bonds with similar calculated distances (2.90–2.93 Å) and interaction energies (between 14.45 and 24.22 kJ mol−1).
As a model for larger cages, B was chosen. In combination with I2F4, a structure is formed in which each cavity of B is occupied by one donor (Fig. 4). The distances between the acceptor sites of B and the incorporated I2F4 are quite large (3.251(9)–3.448(7) Å), and the N⋯I–C binding angles of 142.1(3) and 164.6(3)° indicate that while one imine is bound to the donor, the other contact is rather weak. The encapsulated donor and B show appropriate bond critical points according to QTAIM analysis, and the computed interaction energies reveal the presence of a halogen bond (8.37–13.35 kJ mol−1) and an additional weak interactions (4.85–8.13 kJ mol−1) (Fig. S15 and Table S10, ESI†). The second donor is located on the exterior of B, connecting the two cages via halogen bonding. One of the acceptors is an imine group (N⋯I distance of 2.978(7) Å and N⋯I–C angle of 161.2(3)°), while the π-system of a neighbouring cage B acts as the complementary acceptor (π⋯I distance of 3.281 Å and π⋯I–C angle of 171.32°; distances and angles measured between I and the centre of the nearest π-bond).1h,2 Appropriate bond critical points and a corresponding interaction energy between 14.59 and 22.78 kJ mol−1 were found.
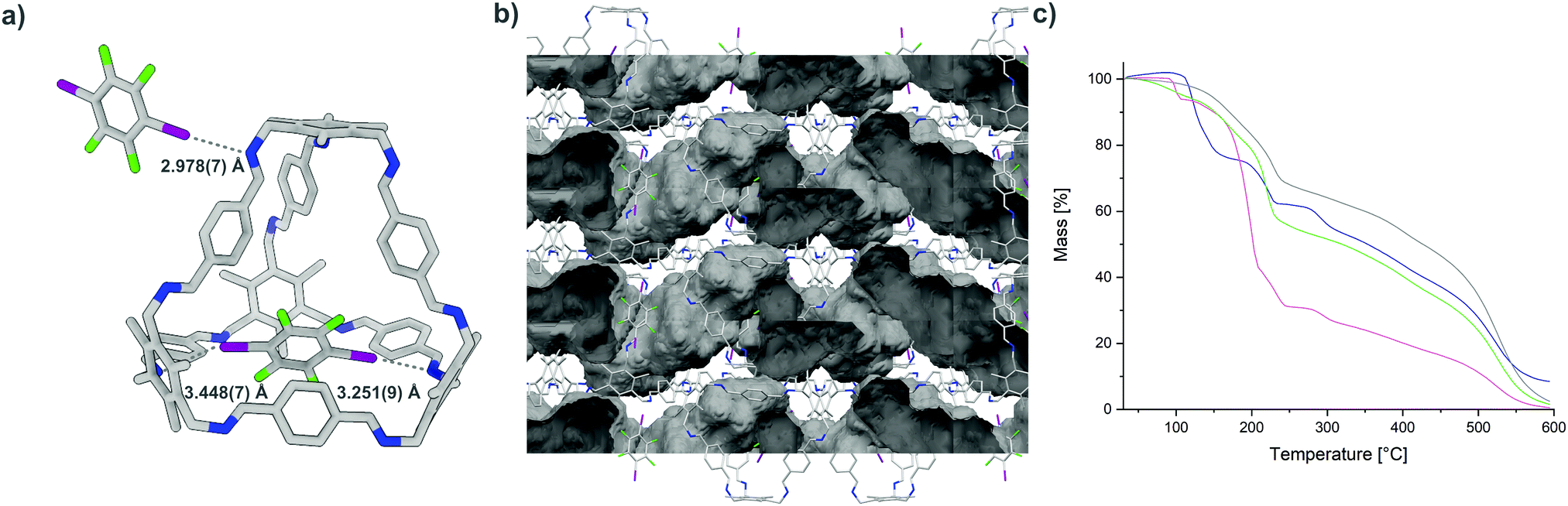 | ||
| Fig. 4 (a) Single-crystal structure of B⋯2(I2F4), N⋯I halogen bond are shown as grey dotted lines; (b) view of B⋯2(I2F4) along the crystallographic c axis, voids (solvent accessible surface area) calculated using a probe of 1.2 Å are depicted in grey colour (highly disordered solvent molecules were removed during the refinement of B⋯2(I2F4), see the ESI† for details); (c) thermogravimetric analysis of A⋯2(I2F4) (blue), A⋯4(I3F3) (pink) and B⋯2(I2F4) obtained by slow evaporation (green) and liquid assisted grinding (grey) (the initial dip for the green curve is due to the loss of remaining solvent, while the sample obtained by grinding (grey curve) contains little to no solvent). | ||
To make the synthesis of our networks more efficient and to enable the synthesis of large quantities, we tested whether the frameworks are also accessible by mechanochemical strategies. Creating co-crystals by neat grinding or liquid-assisted grinding (LAG, also known as solvent-drop grinding or kneading), reduces the amount of solvent needed, as well as the need for its disposal.19 Through mechanochemistry, materials and co-crystals assembled by halogen bonding can also be generated.20 A homogenous powder was obtained in each case after grinding the corresponding donors and acceptors in the desired stoichiometry for 15 minutes at 25 Hz with a small amount of solvent. While the formation of the composites by slow evaporation was usually observable by powder X-ray diffraction (PXRD) analysis, the samples obtained by LAG showed a higher amorphous fraction. As IR spectroscopy has been used previously to observe the formation of halogen-bonded adducts, especially for those obtained by grinding,20b we recorded spectra for the starting materials, as well as for the networks obtained by evaporation and LAG, showing the successful formation. The stability of A⋯2(I2F4), A⋯4(I3F3), and B⋯2(I2F4) was initially investigated by thermogravimetric analysis, showing that the materials obtained by evaporation or LAG are stable to at least 90 °C. Additionally, the different materials were soaked in n-pentane and subsequently kept under high vacuum at 60 °C for 10–16 hours to determine whether they could be activated. The PXRD or IR spectra recorded showed no major decomposition of the networks, even though the highly fluorinated donors are known to sublime under similar conditions (Fig. S34–S37, ESI†) and the 2D pore network of B⋯2(I2F4) is able to take up N2 (Fig. S38 and S39, ESI†).
Herein, we could show that halogen bonding using electron-poor, highly fluorinated donors is an excellent method to post-modify imine-based materials. The novel halogen-bonded frameworks based on POCs can be synthesized by co-crystallisation or LAG. Detailed QTAIM and computed binding analyses in combination with SC-XRD studies gave a comprehensive insight into the previously underexplored halogen bonding of imines. In addition, because SC-XRD analyses of large and porous compounds are inherently difficult due to diffuse diffraction within the reciprocal space at higher resolutions, we believe that this is a viable technique for future analysis of large and very large, novel POC-based compounds using in-house SC-XRD machines after network formation with iodine-containing donors enhancing the scattering. The intermolecular interactions that lead to the formation of the regular crystal lattice can be influenced by employing a reversible and simple approach for binding with the most common functional group used for POC formation, leading to new network topologies and adding a methodology for the development of complex materials for advanced technologies.
This work was funded by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) – SCHM 3101/5-1. We are continuously supported by The Center for Structural Studies, which is funded by the Deutsche Forschungsgemeinschaft (DFG Grant number 417919780) and INST 208/740-1 FUGG. Computational infrastructure and support were provided by the Centre for Information and Media Technology at Heinrich Heine University Düsseldorf. S. H. and K. W. thank the University of Innsbruck for financial support.
Conflicts of interest
There are no conflicts to declare.Notes and references
- (a) R. Kampes, S. Zechel, M. D. Hager and U. S. Schubert, Chem. Sci., 2021, 12, 9275 RSC; (b) Y.-J. Zhu, Y. Gao, M.-M. Tang, J. Rebek and Y. Yu, Chem. Commun., 2021, 57, 1543 RSC; (c) V. Nemec, K. Lisac, N. Bedeković, L. Fotović, V. Stilinović and D. Cinčić, CrystEngComm, 2021, 23, 3063 RSC; (d) D. von der Heiden, A. Vanderkooy and M. Erdélyi, Coord. Chem. Rev., 2020, 407, 213147 CrossRef CAS; (e) L. Turunen and M. Erdélyi, Chem. Soc. Rev., 2020, 49, 2688 RSC; (f) M. Saccone and L. Catalano, J. Phys. Chem. B, 2019, 123, 9281 CrossRef CAS PubMed; (g) C. A. Gunawardana and C. B. Aakeröy, Chem. Commun., 2018, 54, 14047 RSC; (h) G. Cavallo, P. Metrangolo, R. Milani, T. Pilati, A. Priimagi, G. Resnati and G. Terraneo, Chem. Rev., 2016, 116, 2478 CrossRef CAS PubMed; (i) A. Priimagi, G. Cavallo, P. Metrangolo and G. Resnati, Acc. Chem. Res., 2013, 46, 2686 CrossRef CAS PubMed; (j) M. Erdélyi, Chem. Soc. Rev., 2012, 41, 3547 RSC; (k) M. G. Sarwar, B. Dragisic, L. J. Salsberg, C. Gouliaras and M. S. Taylor, J. Am. Chem. Soc., 2010, 132, 1646 CrossRef CAS PubMed.
- G. R. Desiraju, P. S. Ho, L. Kloo, A. C. Legon, R. Marquardt, P. Metrangolo, P. Politzer, G. Resnati and K. Rissanen, Pure Appl. Chem., 2013, 85, 1711 CrossRef CAS.
- (a) E. Uran, L. Fotović, N. Bedeković, V. Stilinović and D. Cinčić, Crystals, 2021, 11, 529 CrossRef CAS; (b) A. J. Peloquin, J. M. McCollum, C. D. McMillen and W. T. Pennington, Angew. Chem., Int. Ed., 2021, 60, 22983 CrossRef CAS PubMed; (c) L. Happonen, J. M. Rautiainen and A. Valkonen, Cryst. Growth Des., 2021, 21, 3409 CrossRef CAS; (d) A. S. Mikherdov, A. S. Novikov, V. P. Boyarskiy and V. Y. Kukushkin, Nat. Commun., 2020, 11, 2921 CrossRef CAS PubMed; (e) Y. Xu, J. Huang, B. Gabidullin and D. L. Bryce, Chem. Commun., 2018, 54, 11041 RSC; (f) R. Núñez, P. Farràs, F. Teixidor, C. Viñas, R. Sillanpää and R. Kivekäs, Angew. Chem., Int. Ed., 2006, 45, 1270 CrossRef PubMed.
- (a) S. Zheng, J. Han, X. Jin, Q. Ye, J. Zhou, P. Duan and M. Liu, Angew. Chem., Int. Ed., 2021, 60, 22711 CrossRef CAS PubMed; (b) S. An, A. Hao and P. Xing, ACS Nano, 2021, 15, 15306 CrossRef CAS PubMed.
- P. M. J. Szell, A. Siiskonen, L. Catalano, G. Cavallo, G. Terraneo, A. Priimagi, D. L. Bryce and P. Metrangolo, New J. Chem., 2018, 42, 10467 RSC.
- (a) E. Nieland, T. Topornicki, T. Kunde and B. M. Schmidt, Chem. Commun., 2019, 55, 8768 RSC; (b) E. Nieland, O. Weingart and B. M. Schmidt, Beilstein J. Org. Chem., 2019, 15, 2013 CrossRef CAS PubMed; (c) T. K. Wijethunga, M. Đaković, J. Desper and C. B. Aakeröy, Acta Crystallogr., 2017, B73, 163 Search PubMed; (d) M. A. Sinnwell and L. R. MacGillivray, Angew. Chem., Int. Ed., 2016, 55, 3477 CrossRef CAS PubMed.
- (a) U. Warzok, M. Marianski, W. Hoffmann, L. Turunen, K. Rissanen, K. Pagel and C. A. Schalley, Chem. Sci., 2018, 9, 8343 RSC; (b) O. Dumele, B. Schreib, U. Warzok, N. Trapp, C. A. Schalley and F. Diederich, Angew. Chem., Int. Ed., 2017, 56, 1152 CrossRef CAS PubMed; (c) L. Turunen, U. Warzok, C. A. Schalley and K. Rissanen, Chemistry, 2017, 3, 861 CrossRef CAS; (d) L. Turunen, A. Peuronen, S. Forsblom, E. Kalenius, M. Lahtinen and K. Rissanen, Chem. – Eur. J., 2017, 23, 11714 CrossRef CAS PubMed; (e) L. Turunen, U. Warzok, R. Puttreddy, N. K. Beyeh, C. A. Schalley and K. Rissanen, Angew. Chem., Int. Ed., 2016, 55, 14033 CrossRef CAS PubMed; (f) N. K. Beyeh, F. Pan and K. Rissanen, Angew. Chem., Int. Ed., 2015, 54, 7303 CrossRef CAS PubMed; (g) O. Dumele, N. Trapp and F. Diederich, Angew. Chem., Int. Ed., 2015, 54, 12339 CrossRef CAS PubMed; (h) C. B. Aakeröy, A. Rajbanshi, P. Metrangolo, G. Resnati, M. F. Parisi, J. Desper and T. Pilati, CrystEngComm, 2012, 14, 6366 RSC.
- (a) G. Gong, S. Lv, J. Han, F. Xie, Q. Li, N. Xia, W. Zeng, Y. Chen, L. Wang, J. Wang and S. Chen, Angew. Chem., Int. Ed., 2021, 60, 14831 CrossRef CAS PubMed; (b) J. Zhou, L. Stojanović, A. A. Berezin, T. Battisti, A. Gill, B. M. Kariuki, D. Bonifazi, R. Crespo-Otero, M. R. Wasielewski and Y.-L. Wu, Chem. Sci., 2021, 12, 767 RSC; (c) N. Chongboriboon, K. Samakun, T. Inprasit, F. Kielar, W. Dungkaew, L. W.-Y. Wong, H. H.-Y. Sung, D. B. Ninković, S. D. Zarić and K. Chainok, CrystEngComm, 2020, 22, 24 RSC; (d) I. G. Grosu, L. Pop, M. Miclǎuş, N. D. Hǎdade, A. Terec, A. Bende, C. Socaci, M. Barboiu and I. Grosu, Cryst. Growth Des., 2020, 20, 3429 CrossRef CAS; (e) V. I. Nikolayenko, D. C. Castell, D. P. van Heerden and L. J. Barbour, Angew. Chem., Int. Ed., 2018, 57, 12086 CrossRef CAS PubMed; (f) M. C. Pfrunder, A. J. Brock, J. J. Brown, A. Grosjean, J. Ward, J. C. McMurtrie and J. K. Clegg, Chem. Commun., 2018, 54, 3974 RSC; (g) S. Shankar, O. Chovnik, L. J. W. Shimon, M. Lahav and M. E. van der Boom, Cryst. Growth Des., 2018, 18, 1967 CrossRef CAS.
- (a) T. Kunde, T. Pausch and B. M. Schmidt, Eur. J. Org. Chem., 2021, 5844 CrossRef CAS; (b) M. A. Little and A. I. Cooper, Adv. Funct. Mater., 2020, 30, 1909842 CrossRef CAS; (c) M. Mastalerz, Acc. Chem. Res., 2018, 51, 2411 CrossRef CAS PubMed; (d) F. Beuerle and B. Gole, Angew. Chem., Int. Ed., 2018, 57, 4850 CrossRef CAS PubMed.
- M. J. Bojdys, M. E. Briggs, J. T. A. Jones, D. J. Adams, S. Y. Chong, M. Schmidtmann and A. I. Cooper, J. Am. Chem. Soc., 2011, 133, 16566 CrossRef CAS PubMed.
- (a) Q. Zhu, X. Wang, R. Clowes, P. Cui, L. Chen, M. A. Little and A. I. Cooper, J. Am. Chem. Soc., 2020, 142, 16842 CrossRef CAS PubMed; (b) J.-X. Ma, J. Li, Y.-F. Chen, R. Ning, Y.-F. Ao, J.-M. Liu, J. Sun, D.-X. Wang and Q.-Q. Wang, J. Am. Chem. Soc., 2019, 141, 3843 CrossRef CAS PubMed.
- T. Hasell, J. L. Culshaw, S. Y. Chong, M. Schmidtmann, M. A. Little, K. E. Jelfs, E. O. Pyzer-Knapp, H. Shepherd, D. J. Adams, G. M. Day and A. I. Cooper, J. Am. Chem. Soc., 2014, 136, 1438–1448 CrossRef CAS PubMed.
- (a) X. Liu and P. H. Toy, Adv. Synth. Catal., 2020, 362, 3437 CrossRef CAS; (b) T. Suzuki, S. Kuwano and T. Arai, Adv. Synth. Catal., 2020, 362, 3208 CrossRef CAS; (c) M. Kaasik, A. Metsala, S. Kaabel, K. Kriis, I. Järving and T. Kanger, J. Org. Chem., 2019, 84, 4294 CrossRef CAS PubMed; (d) R. Haraguchi, S. Hoshino, M. Sakai, S. Tanazawa, Y. Morita, T. Komatsu and S. Fukuzawa, Chem. Commun., 2018, 54, 10320 RSC; (e) Y. Takeda, D. Hisakuni, C.-H. Lin and S. Minakata, Org. Lett., 2015, 17, 318 CrossRef CAS PubMed; (f) J.-L. Syssa Magalé, K. Boubekeur, P. Palvadeau, A. Meerschaut and B. Schöllhorn, J. Mol. Struct., 2004, 691, 79 CrossRef.
- V. Santolini, M. Miklitz, E. Berardo and K. E. Jelfs, Nanoscale, 2017, 9, 5280 RSC.
- R. L. Greenaway, V. Santolini, M. J. Bennison, B. M. Alston, C. J. Pugh, M. A. Little, M. Miklitz, E. G. B. Eden-Rump, R. Clowes, A. Shakil, H. J. Cuthbertson, H. Armstrong, M. E. Briggs, K. E. Jelfs and A. I. Cooper, Nat. Commun., 2018, 9, 2849 CrossRef CAS PubMed.
- (a) M. Müller, M. Albrecht, V. Gossen, T. Peters, A. Hoffmann, G. Raabe, A. Valkonen and K. Rissanen, Chem. – Eur. J., 2010, 16, 12446 CrossRef PubMed; (b) K. Xu, D. M. Ho and R. A. Pascal, J. Am. Chem. Soc., 1994, 116, 105 CrossRef CAS.
- (a) T. D. Kühne, M. Iannuzzi, M. Del Ben, V. V. Rybkin, P. Seewald, F. Stein, T. Laino, R. Z. Khaliullin, O. Schütt, F. Schiffmann, D. Golze, J. Wilhelm, S. Chulkov, M. H. Bani-Hashemian, V. Weber, U. Borštnik, M. Taillefumier, A. S. Jakobovits and A. Lazzaro, et al. , J. Chem. Phys., 2020, 152, 194103 CrossRef PubMed; (b) P. Giannozzi, S. Baroni, N. Bonini, M. Calandra, R. Car, C. Cavazzoni, D. Ceresoli, G. L. Chiarotti, M. Cococcioni, I. Dabo, A. Dal Corso, S. de Gironcoli, S. Fabris, G. Fratesi, R. Gebauer, U. Gerstmann, C. Gougoussis, A. Kokalj, M. Lazzeri, L. Martin-Samos, N. Marzari, F. Mauri, R. Mazzarello, S. Paolini, A. Pasquarello, L. Paulatto, C. Sbraccia, S. Scandolo, G. Sclauzero, A. P. Seitsonen, A. Smogunov, P. Umari and R. M. Wentzcovitch, J. Phys.: Condens. Matter, 2009, 21, 395502 CrossRef PubMed.
- T. Lu and F. Chen, J. Comput. Chem., 2012, 33, 580 CrossRef CAS PubMed.
- T. Friščić, Chem. Soc. Rev., 2012, 41, 3496–3510 RSC.
- (a) M. Arhangelskis, F. Topić, P. Hindle, R. Tran, A. J. Morris, D. Cinčić and T. Friščić, Chem. Commun., 2020, 56, 8293 RSC; (b) C. Aakeröy, M. Baldrighi, J. Desper, P. Metrangolo and G. Resnati, Chem. – Eur. J., 2013, 19, 16240 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2115251–2115254. For ESI and crystallographic data in CIF or other electronic format see https://doi.org/10.1039/d2cc00799a |
| This journal is © The Royal Society of Chemistry 2022 |