
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Transition structures for the oxy–ene reaction†
Shengwen
Yang
ab,
Solène
Miaskiewicz
a,
Christophe
Bour
 a,
Aurélien
Alix
a and
Vincent
Gandon
*ab
a,
Aurélien
Alix
a and
Vincent
Gandon
*ab
aUniversité Paris-Saclay, CNRS, Institut de Chimie Moléculaire et des Matériaux d'Orsay, 91405, Orsay, France. E-mail: vincent.gandon@universite-paris-saclay.fr
bLaboratoire de Chimie Moléculaire (LCM), CNRS UMR 9168, Ecole Polytechnique, Institut Polytechnique de Paris, Route de Saclay, 91128 Palaiseau cedex, France
First published on 21st March 2022
Abstract
An overlooked pericyclic reaction between allyl alcohols and alkenes to form carbonyl compounds is analyzed. It combines the characteristic features of the Alder–ene reaction and of the oxy-Cope rearrangement. This oxy–ene reaction could be involved in biosynthetic pathways.
Discovered in 1943,1 the Alder–ene reaction is a fundamental organic transformation that keeps receiving considerable attention (Scheme 1A).2 The computational study of Fernández and Bickelhaupt has explained why it usually requires a high temperature despite its pericyclic nature.3 The difficult deformation of the reactants to reach the geometry of the aromatic six-membered transition state leads to a high activation strain and consequently a high barrier. However, the use of polarized enes and enophiles greatly reduces the activation energies and allows Alder–ene reactions to take place under gentle heating, or even at room temperature.4
 | ||
| Scheme 1 Some pericyclic reactions. | ||
Polarized Alder–ene reactions are believed to occur naturally.5 The first enzyme-catalyzed Alder–ene reaction reported in 2020 by Zhou, Houk, Tang and co-workers6 supports this hypothesis. This breakthrough could lead to new applications of the Alder–ene reaction and encourages the reconsideration of biosynthetic routes towards some natural products. In that respect, the biogenesis of the aristotelia alkaloid aristone7 has long been debated (Scheme 2).8 After ruling out an earlier hypothesis,8a Borschberg rationalized the formation of aristone and its stereochemistry by an intramolecular [1,5]-H shift from intermediate 1, which displays an allyl alcohol and an indole moiety.8b,c This concerted intramolecular process would lead to enol 2 and then aristone after keto-enol tautomerism. Because of its similarity with the Alder–ene reaction and with the oxy-Cope rearrangement in terms of reactants and products (Scheme 1B), Borschberg referred to this transformation as “oxyanion-ene” reaction in 1996.8c
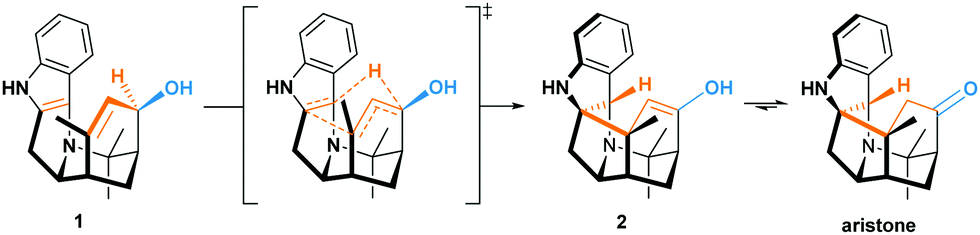 | ||
| Scheme 2 Borschberg's hypothesis regarding the biosynthesis of aristone. | ||
In fact, such a reaction had been described in 1981 by Klärner and co-workers who named it “oxy-homodienyl hydrogen shift” by analogy with the oxy-Cope rearrangement.9 The stereoselective thermolysis of (Z)-hexa-2,5-dienols like compound 3 into cis-cyclopropane 4 was achieved at ca 260 °C, overcoming an experimentally determined Gibbs free energy of activation of 43.5 kcal mol−1 (Scheme 3). Borschberg designed his own substrates such as 5 and investigated their thermally-induced reaction, coined “oxy–ene” in 2001 (Scheme 1C),10 pointing out that stereoselective transformations could be achieved at high temperature. It is well-known that the oxy-Cope rearrangement can be accelerated by deprotonation of the hydroxy group.11 This has been attributed to the bond-weakening effect induced by the alkoxide to the adjacent C3–C4 bond.12 In a second 2001 paper, Borschberg reported anionic oxy–ene reactions, such as 5 → 8 → 9 → 7, which proved to be faster than neutral ones.13 Of note, the first anionic oxy retro–ene reaction was reported by Jung and Davidov the same year.14
 | ||
| Scheme 3 Reported neutral and anionic oxy–ene reactions. | ||
The high energy demand of the reported oxy–ene reactions seems to question the viability of such a process in a biological environment. However, the enophilic moiety of compound 1 is quite specific, being both an indole and an enamine double bond. This substrate also presents acidic and basic sites that could form strong hydrogen bonds. In 1998, Barrero and co-workers reported that, when heated in toluene, (+)-salonitenolide 10 undergoes thermal Cope rearrangement into (+)-dehydromelitensin 11 (Scheme 4).15 In the presence of 0.02 equiv of PdCl2(PhCN)2, 10 transforms into a mixture of 11 and (+)-stoebenolide 12. The latter can be obtained selectively when using an equimolar amount of the Pd(II) complex. With a low concentration of Pd(II), ring opening leads to 11 as in a typical Lewis acid catalyzed formal Cope rearrangement.16 At higher Pd(II) concentration, the ligand acts as a base, leading to 12 as formal oxy–ene product.
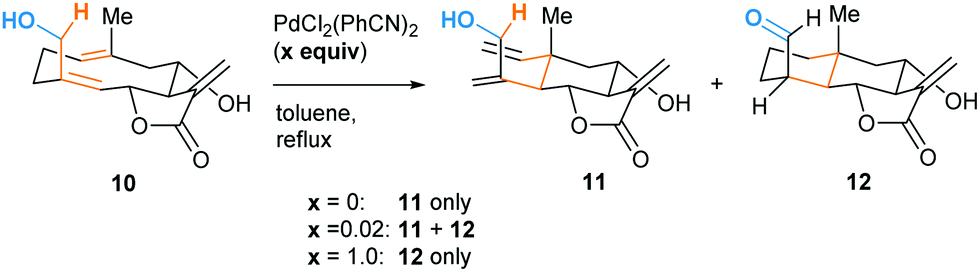 | ||
| Scheme 4 Pd(II)-promoted rearrangement of germacranolides: formal Cope (11) and oxy–ene (12) reactions. | ||
The capacity of enzymes to catalyze Alder–ene reactions6 offers a new perspective on biosynthetic pathways. We report herein the first theoretical analysis of the oxy–ene reaction under neutral or basic conditions using parent compounds. We then shed light on its possible role in the biosynthesis of aristone using enzyme residues (theozymes). We started our investigations with the parent system A composed of allyl alcohol as ene partner and ethene as enophile (Fig. 1, see the ESI† for computational details). A concerted pathway leading to enol ether B was modelled. The Gibbs free energy of activation to reach TSAB is 42.1 kcal mol−1, which is consistent with the experimental value of 43.5 kcal mol−1 reported by Klärner et al. for the intramolecular cyclization of (Z)-hexa-2,5-dienol.9 Of note, A was taken in its s-cis conformation, leading to a cis enol. With the s-trans conformation, a slightly higher barrier of 42.3 kcal mol−1 was obtained. With substituted allyl alcohols (vide infra), the s-trans conformation is the one that leads to the lowest barrier.
 | ||
| Fig. 1 Energy profiles at 483.15 K (kcal mol−1) and transition state geometries (selected distances in Å) of the neutral (A to B) and anionic (C to D) oxy–ene reactions of allyl alcohol with ethene computed at the ωB97XD/def2-QZVPP//ωB97XD/6-311+G(d,p) level. | ||
The transition state of a pericyclic reaction is expected to be aromatic. To confirm this property, we computed the nuclear independent chemical shift (NICS(0)) value17 at the center of the six-membered ring, using the GIAO method18 at the ωB97XD/6-311+G(d,p) level of theory (Table 1). We obtained -24.7 ppm for TSAB, which is very close from the parent aromatic Alder-ene transition state (−24.2 ppm at our level of theory).3
| TS | NICS(0) | TS | NICS(0) | TS | NICS(0) |
|---|---|---|---|---|---|
| a With all additives. | |||||
| TSAB | −24.7 | TSGH | −13.4 | TS12 | −20.5 |
| TSCD | −16.6 | TSMN | −13.6 |

|
−20.1 |
We then performed an activation-strain analysis of the transition state according to the general equation  , where ΔEstrain is the strain energy and ΔEint the instantaneous interaction energy (Table 2).19 The results are very close from the parent Alder–ene reaction for which the barrier mostly stems from the activation strain of the ene partner.3 To reach the geometry of the aromatic transition state, an energy of 32.1 kcal mol−1 is required to deform allyl alcohol but only 11.6 kcal mol−1 to deform ethene. As the electronic interaction between the strained reactants is just −11.7 kcal mol−1, the energy of activation remains as high as 33.0 kcal mol−1.
, where ΔEstrain is the strain energy and ΔEint the instantaneous interaction energy (Table 2).19 The results are very close from the parent Alder–ene reaction for which the barrier mostly stems from the activation strain of the ene partner.3 To reach the geometry of the aromatic transition state, an energy of 32.1 kcal mol−1 is required to deform allyl alcohol but only 11.6 kcal mol−1 to deform ethene. As the electronic interaction between the strained reactants is just −11.7 kcal mol−1, the energy of activation remains as high as 33.0 kcal mol−1.
 .a
.a
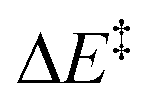


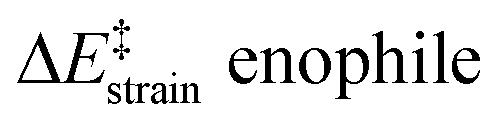
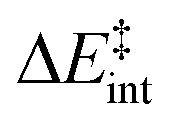
Thus, with simple substrates, the oxy–ene reaction has the same characteristic as the Alder–ene reaction. However, the presence of the OH group allows the formation of an alkoxy anion by deprotonation, which could have a dramatic impact on the reaction barrier by analogy with the oxy-Cope rearrangement. The free energy of activation indeed drops to 25.5 kcal mol−1 in the alkoxide series (Fig. 1, C to D). The striking geometrical difference between TSCD and TSAB is the much longer distance of the forming C–C bond (2.93 vs. 2.16 Å). The aromatic character of the transition state is also less pronounced (Table 1, NICS(0) −16.6 ppm). Compared to the alcohol, the alkoxide is easier to deform to reach the geometry of the transition state, as shown by the activation-strain analysis (Tables 2, 18.3 vs. 32.1 kcal mol−1). Thus, the transition state is more reactant-like. This phenomenon can be explained by the weakening of the allylic C–H bond in the alkoxide series. The homolytic bond dissociation energy (BDE) of this bond in allyl alcohol is ΔE 83.4 kcal mol−1. In the corresponding alkoxide, the BDE becomes ΔE 57.2 kcal mol−1 only. This effect is consistent with that reported for the oxy-Cope rearrangement.12a
Intramolecular reactions corresponding to Klärner's and Borschberg's substrates were then studied, which confirmed their pericyclic nature and the lowering of the cyclization barrier after deprotonation of the alcohol (see the ESI,† Fig S1, compounds E to L′), although there is a striking difference between the naked alkoxide and the metallated one.
The weakening of the allylic C–H bond by deprotonation is a good lead to rationalize the formation of aristone under biological conditions (Scheme 2). So far, all barriers computed with neutral substrates (38.3–42.1 kcal mol−1) are incompatible with natural processes, and the calculated route to aristone makes no exception (Fig. 2). The transition state connecting 1 to 2 lies 42.3 kcal mol−1 above the former. On the other hand, if the hydroxy group is deprotonated, the transition state is located only 23.6 kcal mol−1 above the allyl alkoxide (M).
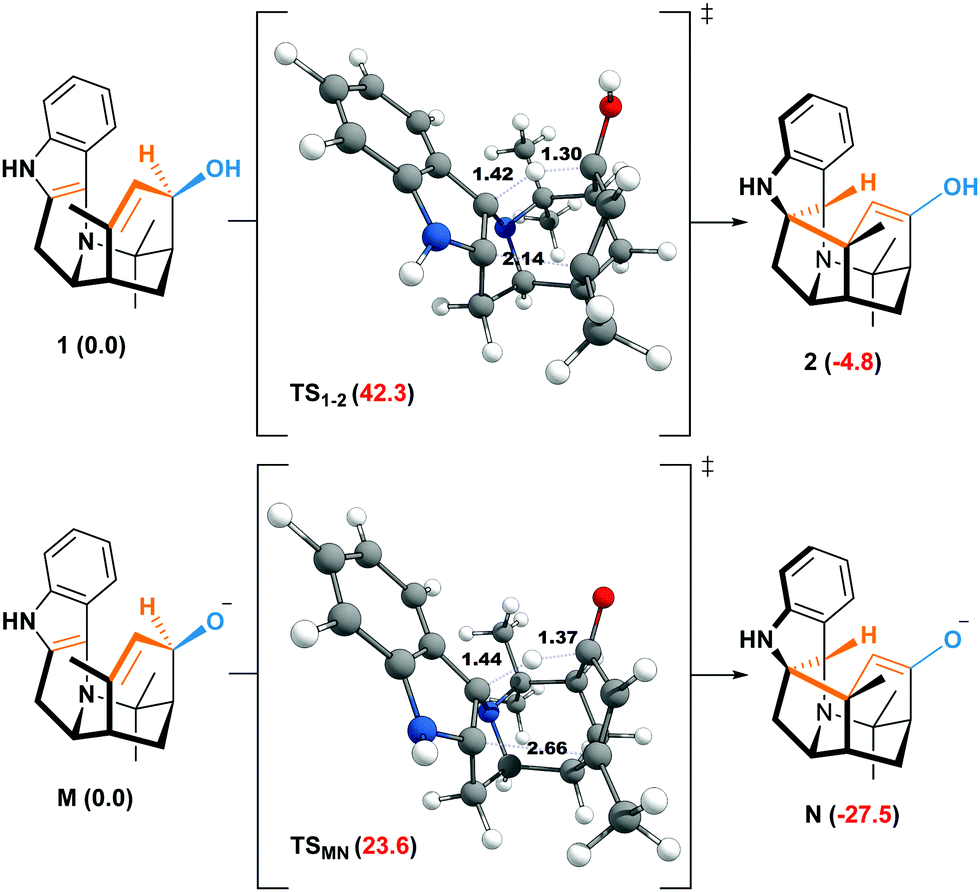 | ||
| Fig. 2 Free energies at 298.15 K (ΔG kcal mol−1) of the neutral and anionic oxy–ene reactions corresponding to Scheme 2. | ||
In vivo, biocatalysts are able to stabilize transition states or intermediates and to reduce the activation energy.20 For example, the accepted mechanism for zinc-dependent alcohol dehydrogenase enzymes first supposes that the alcohol substrate binds to the active site by coordination to the zinc ion. Then, deprotonation of the hydroxy group through a proton transfer to the solvent via a serine-NAD(P)+ cofactor-histidine triad allows for the formation of a Zn2+-stabilized alkoxide intermediate.21 Finally, a hydride transfer from the carbon of the alkoxide to the cofactor completes the reaction and releases the corresponding aldehyde or ketone.22 In such scenario, the in vivo deprotonation of a secondary alcohol such as 1 could be effective.
Even if not deprotonated, the formation of a H-bond network between the hydroxy group and amino acid residues of the active site of a biocatalyst might also weaken the allylic C–H bond. Besides, H-bonding at the tertiary amine moiety of the transition state could influence the reaction rate. In the case of the enzyme-catalyzed Alder–ene reaction, Zhou, Houk, Tang and co-workers6 used the theozyme23 computational approach to rationalize the acceleration. A lysine residue was simplified as MeNH3+ and histidine as imidazole. While in our case the biocatalyst is not known, we wanted to verify whether typical H-bond donors or acceptors used in theozyme computations could influence the reaction barrier (Fig. 3). With MeNH3+, we observed the protonation of the tertiary amine during optimization. Methylamine remained H-bonded to the newly formed ammonium 2*. This protonation lowered the energy of the oxy–ene transition state  by 4.4 kcal mol−1 compared to TS1-2 (from 42.3 to 37.9 kcal mol−1). Further 5.2 kcal mol−1 were gained with imidazole H-bonded to the hydroxy group (32.7 kcal mol−1). Of note, activation of the two sites is important, as using only imidazole raised the barrier to 38.7 kcal mol−1. As typical abzyme model,23 we then used HCO2− and MeOH as H-bond acceptor and donor respectively.24 This combination had a dramatic impact on the barrier, which was lowered to 30.1 kcal mol−1, i.e. 12.2 kcal mol−1 less than the initial value. Interestingly, the carboxylate bridges the OH and NH groups. As witnessed by the removal of MeOH, activation of the two sites was again more efficient. Replacing MeOH by HCO2H further reduced the energy of the transition state to 29.3 kcal mol−1. Thus, addition of amino acid residues strongly decreases the oxy–ene barrier. This effect is still valid if the OH group is fully deprotonated. In this case, the barrier becomes 19.1 kcal mol−1 (vs. 23.6 kcal mol−1 in Fig. 2) with one H-bonded HCO2H
by 4.4 kcal mol−1 compared to TS1-2 (from 42.3 to 37.9 kcal mol−1). Further 5.2 kcal mol−1 were gained with imidazole H-bonded to the hydroxy group (32.7 kcal mol−1). Of note, activation of the two sites is important, as using only imidazole raised the barrier to 38.7 kcal mol−1. As typical abzyme model,23 we then used HCO2− and MeOH as H-bond acceptor and donor respectively.24 This combination had a dramatic impact on the barrier, which was lowered to 30.1 kcal mol−1, i.e. 12.2 kcal mol−1 less than the initial value. Interestingly, the carboxylate bridges the OH and NH groups. As witnessed by the removal of MeOH, activation of the two sites was again more efficient. Replacing MeOH by HCO2H further reduced the energy of the transition state to 29.3 kcal mol−1. Thus, addition of amino acid residues strongly decreases the oxy–ene barrier. This effect is still valid if the OH group is fully deprotonated. In this case, the barrier becomes 19.1 kcal mol−1 (vs. 23.6 kcal mol−1 in Fig. 2) with one H-bonded HCO2H  . Although this low value of 19.1 kcal mol−1 represents a lower limit as it is obtained with a naked alkoxide (see the discussion in the ESI† regarding the deprotonation), it seems conceivable that an oxy–ene reaction occurs in vivo in the presence of many H-bond donor or acceptor residues.
. Although this low value of 19.1 kcal mol−1 represents a lower limit as it is obtained with a naked alkoxide (see the discussion in the ESI† regarding the deprotonation), it seems conceivable that an oxy–ene reaction occurs in vivo in the presence of many H-bond donor or acceptor residues.
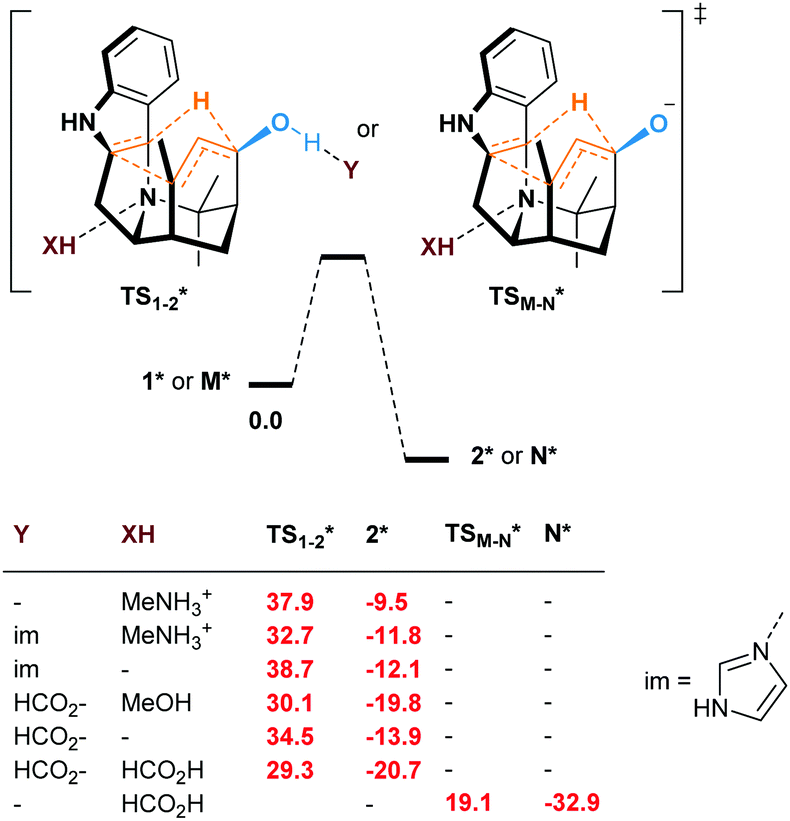 | ||
| Fig. 3 Free energies at 298.15 K (ΔG kcal mol−1) of the oxy–ene reaction of 1* or M* in the presence of theozyme residues (* = in the presence of Y and/or XH). | ||
Having much in common with the Alder–ene reaction and some similarities with the oxy-Cope rearrangement, the oxy–ene is an overlooked pericyclic reaction. Just like the Alder–ene, the oxy–ene could play a significant role in biological processes and become a useful tool in synthetic organic chemistry. Until now, information about the oxy–ene reaction has remained scattered in the literature. This study puts together the pieces of the puzzle and provides the theoretical background of this underexplored subfield of pericyclic group transfer reactions.
S. Y. thanks the CSC for PhD grant. We thank the support of UPSaclay, Ecole Polytechnique, ANR-18-CE07-0033-01 (HICAT) and GENCI (2020-A0070810977).
Conflicts of interest
There are no conflicts of interest to declare.Notes and references
- (a) K. Alder, F. Pascher and A. Schmitz, Ber. Dtsch. Chem. Ges. B, 1943, 76, 27 CrossRef; (b) K. Alder and T. Noble, Ber. Dtsch. Chem. Ges. B, 1943, 76, 54 CrossRef; (c) K. Alder and C.-H. Schmidt, Ber. Dtsch. Chem. Ges. B, 1943, 76, 183 CrossRef; (d) H. M. R. Hoffmann, Angew. Chem., Int. Ed. Engl., 1969, 8, 556 CrossRef CAS.
- See inter alia: (a) S. Gupta, Y. Lin, Y. Xia, D. J. Wink and D. Lee, Chem. Sci., 2019, 10, 2212 RSC; (b) X. Chen, Y. Lu, Z. Guan, L. Gu, C. Chen, H. Zhu, Z. Luo and Y. Zhang, Org. Lett., 2021, 23, 3173 CrossRef CAS PubMed.
- I. Fernández and F. M. Bickelhaupt, J. Comput. Chem., 2012, 33, 509 CrossRef PubMed.
- (a) L. R. Domingo, M. J. Aurell and P. Pérez, Org. Biomol. Chem., 2014, 12, 7581 RSC; (b) T. J. J. Müller,Comprehensive Organic Synthesis II, 2014, vol. 5, ch. 5.01, pp. 1–65 Search PubMed.
- A. W. Jensen, D. K. Mohanty and W. L. Dilling, Bioorg. Med. Chem., 2019, 27, 686 CrossRef CAS PubMed.
- M. Ohashi, C. S. Jamieson, Y. Cai, D. Tan, D. Kanayama, M.-C. Tang, S. M. Anthony, J. V. Chari, J. S. Barber, E. Picazo, T. B. Kakule, S. Cao, N. K. Garg, J. Zhou, K. N. Houk and Y. Tang, Nature, 2020, 586, 64 CrossRef CAS PubMed.
- H. R. Arias, M. O. Ortells, D. Feuerbach, V. Burgos and C. Paz, J. Nat. Prod., 2019, 82, 1953 CrossRef CAS PubMed.
- (a) V. Zabel, W. H. Watson, M. Bittner and M. Silva, J. Chem. Soc., Perkin Trans. 1, 1980, 2842 RSC; (b) H.-J. Borschberg, in The Chemistry of Heterocyclic Compounds, Supplement to Part 4, Vol. 25, ed. J. E. Saxton, Wiley, Chichester, 1994, pp. 15–56 Search PubMed; (c) H.-J. Borschberg, in The Alkaloids: Chemistry and Pharmacology, ed. G. A. Cordell, Academic Press, New York, 1996, vol. 48, pp. 191–248 Search PubMed.
- F.-G. Klärner, W. Rüngeler and W. Maifeld, Angew. Chem., Int. Ed. Engl., 1981, 20, 595 CrossRef.
- G. A. Schmid and H.-J. Borschberg, Helv. Chim. Acta, 2001, 84, 388 CrossRef CAS.
- (a) D. A. Evans and A. M. Golob, J. Am. Chem. Soc., 1975, 97, 4765 CrossRef CAS; (b) D. A. Evans, D. J. Baillargeon and J. V. Nelson, J. Am. Chem. Soc., 1978, 100, 2242 CrossRef CAS.
- (a) M. L. Steigerwald, W. A. Goddard III and D. A. Evans, J. Am. Chem. Soc., 1979, 101, 1994 CrossRef CAS; (b) H. Y. Yoo, K. N. Houk, J. K. Lee, M. A. Scialdone and A. I. Meyers, J. Am. Chem. Soc., 1998, 120, 205 CrossRef CAS; (c) S. M. Schulze, N. Santella, J. J. Grabowski and J. K. Lee, J. Org. Chem., 2001, 66, 7247 CrossRef CAS PubMed.
- G. A. Schmid and H.-J. Borschberg, Helv. Chim. Acta, 2001, 84, 401 CrossRef CAS.
- M. E. Jung and P. Davidov, Org. Lett., 2001, 3, 3025 CrossRef CAS PubMed.
- A. F. Barrero, J. E. Oltra and M. Alvarez, Tetrahedron Lett., 1998, 39, 1401 CrossRef CAS.
- L. E. Overman and A. F. Renaldo, J. Am. Chem. Soc., 1990, 112, 3945 CrossRef CAS.
- (a) P. V. R. Schleyer, C. Maerker, A. Dransfeld, H. Jiao and N. J. R. V. Eikema Hommes, J. Am. Chem. Soc., 1996, 118, 6317 CrossRef CAS PubMed; (b) Z. Chen, C. S. Wannere, C. Corminboeuf, R. Puchta and P. V. R. Schleyer, Chem. Rev., 2005, 105, 3842 CrossRef CAS PubMed.
- K. Wolinski, J. F. Hilton and P. Pulay, J. Am. Chem. Soc., 1990, 112, 8251 CrossRef CAS.
- F. M. Bickelhaupt and K. N. Houk, Angew. Chem., Int. Ed., 2017, 56, 10070 CrossRef CAS PubMed.
- (a) L. Pauling, Chem. Eng. News, 1946, 24, 1375 CrossRef CAS; (b) L. Pauling, Nature, 1948, 161, 707 CrossRef CAS PubMed; (c) A. Fersht, Enzyme Structure and Mechanism, W. H. Freeman and Co, New York, 1985 Search PubMed; (d) A. Warshel, P. K. Sharma, M. Kato, Y. Xiang, H. Liu and M. H. M. Olsson, Chem. Rev., 2006, 106, 3210 CrossRef CAS PubMed.
- (a) M. Hennecke and B. V. Plapp, Biochemistry, 1983, 22, 3721 CrossRef CAS PubMed; (b) B. V. Plapp, B. R. Savarimuthu, D. J. Ferraro, J. K. Rubach, E. N. Brown and S. Ramaswamy, Biochemistry, 2017, 56, 3632 CrossRef CAS PubMed.
- Computational studies support this mechanism: (a) P. K. Agarwal, S. P. Webb and S. Hammes-Schiffer, J. Am. Chem. Soc., 2000, 122, 4803 CrossRef CAS; (b) Q. Cui, M. Elstner and M. Karplus, J. Phys. Chem. B, 2002, 106, 2721 CrossRef CAS; (c) C. Lee, D. L. Bedgar, L. B. Davin and N. G. Lewis, Org. Biomol. Chem., 2013, 11, 1127 RSC; (d) S. Moa and F. Himo, J. Inorg. Biochem., 2017, 175, 259 CrossRef CAS PubMed.
- D. J. Tantillo, J. Chen and K. N. Houk, Curr. Opin. Chem. Biol., 1998, 2, 743 CrossRef CAS PubMed.
- Of note, the use of an autoclave washed with soap resulted in an acceleration of the reaction, see: C. Schlicht, PhD thesis, Diss. ETH No. 16341, 2005.
Footnote |
| † Electronic supplementary information (ESI) available: Computational details, additional computations, coordinates and energies. See DOI: 10.1039/d2cc00687a |
| This journal is © The Royal Society of Chemistry 2022 |