
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSilyl formates as hydrosilane surrogates for the transfer hydrosilylation of ketones†
R. Martin
Romero
 ,
Neethu
Thyagarajan
,
Nora
Hellou
,
Clément
Chauvier
,
Timothé
Godou
,
Lucile
Anthore-Dalion
and
Thibault
Cantat
*
,
Neethu
Thyagarajan
,
Nora
Hellou
,
Clément
Chauvier
,
Timothé
Godou
,
Lucile
Anthore-Dalion
and
Thibault
Cantat
*
Université Paris-Saclay, CEA, CNRS, NIMBE, 91191 Gif-sur-Yvette, France. E-mail: thibault.cantat@cea.fr
First published on 3rd May 2022
Abstract
A transfer hydrosilylation of ketones employing silyl formates as hydrosilane surrogates under mild conditions is presented. A total of 24 examples of ketones have been successfully converted to their corresponding silyl ethers with 61–99% yields in the presence of a PNHP-based ruthenium catalyst and silyl formate reagent. The crucial role of the ligand for the transformation is demonstrated.
Catalytic hydrosilylation is a convenient method to reduce carbonyl compounds, providing access to alcohols via silyl ether intermediates.1 The latter are also an important class of protecting groups for alcohols. Their direct synthesis from the corresponding ketone is hence valuable. Transfer hydrosilylation has emerged as an alternative process for this transformation,2 avoiding the use of difficult to handle hydrosilanes, such as the gaseous Me3SiH. This concept was pioneered by Studer3 and Oestreich,4 who reported silicon-substituted cyclohexa-1,4-dienes for the transfer hydrosilylation of alkenes and carbonyl derivatives through radical and ionic processes, respectively (Scheme 1A). The formation of hydrosilylation products is accompanied by the production of quantitative arene derivatives as by-products.
 | ||
| Scheme 1 (A) Hydrosilane surrogates. (B) Applications of silyl formates as hydrosilane surrogates. (C) Ruthenium-catalyzed transfer hydrosilylation of ketones (this work). | ||
We have reported an alternative using silyl formates as renewable liquid surrogates of hydrosilanes, whose only by-product is gaseous CO2.5 The recyclability of these reagents is ensured since they are synthesized in excellent yields from formic acid, a reagent readily available from biomass6 or carbon dioxide.7
Silylformates were initially employed as hydrosilane surrogates in alcohol silylation with iron-8 or ruthenium-based catalysts.9 Transfer hydrosilylation of aldehydes was successfully developed using the Ru-triphos catalyst 1 (Scheme 1B).5 During these transformations, the metal-mediated silyl formate decarboxylation generates a metal hydride species that will provide a metal–alkoxide intermediate upon reaction with the substrate. The final silylation step provides the desired product, closing the catalytic cycle. Interestingly, we could show that silyl hydride species are never formed along this process. Unfortunately, these protocols were ineffective towards the reduction of ketones. In this case, it seems that the steric hindrance around the metal–alkoxide intermediate hampers the final silylation step.5
In order to increase the nucleophilicity of the oxygen atom, we envisioned the possibility of weakening the ruthenium–alkoxide interaction through the action of a cooperative ligand, able to develop H-bonds. We chose the PNHP–ruthenium catalyst 2 that bears a well-known ligand for its participation in metal-catalyzed reactions through his N–H bond.10 Major contributions on complexes bearing PNHP ligands were achieved by Milstein,11 Beller,10b,12 Gusev,13 and Kuriyama.14 These species were successfully applied to the reduction of challenging substrates such as esters or amides.10b,14,15 However, beyond hydrogenation, the use of participative PNHP ligand-based catalysts in hydrosilylation is scarce,16 and, to the best of our knowledge, it was never reported in transfer hydrosilylation reactions.
To test our hypothesis, acetophenone (3a) was submitted for reaction with triethylsilyl formate (5a) and Ru-triphos catalyst 1 in acetonitrile at 90 °C, classical conditions for the transfer hydrosilylation of aldehydes. Under these conditions, no conversion was observed (Table 1, entry 1). Changing catalyst 1 to Ru–PNHP catalyst 2 provided silyl ether 4a in 78% yield (Table 1, entry 2). While substituting CD3CN with d2-dichloromethane completely suppresses the reactivity (Table 1, entry 3), the use of d8-THF, d8-toluene or d6-benzene increased the yields to 99%, 92% and 99%, respectively (Table 1, entries 4–6). Performing the reaction in more environment-friendly solvents such as EtOAc or anisole allowed also the obtention of the product in 97% and 77% yields, respectively (Table 1, entries 7 and 8). Among them, we finally selected d6-benzene to rapidly evaluate the applicability of the reaction due to a lower reaction time (1.5 h). Reducing the catalyst loading from 3 mol% to 1.5 mol% results in a drop of yield to 79% (Table 1, entry 9). Decreasing the temperature to 50 °C increases the required reaction time (36 h) to obtain a comparable yield of the silylated alcohol 4a (99%) (Table 1, entry 10).
|
|
|||||
|---|---|---|---|---|---|
| Entry | Catalyst (mol%) | Solvent | T (°C) | t (h) | Yieldb (%) |
| a 0.1 mmol scale. b Yields are determined by 1H NMR with mesitylene as an internal standard. See ESI for more details. | |||||
| 1 | 1 (3) | CD3CN | 90 | 24 | 0 |
| 2 | 2 (3) | CD3CN | 90 | 11 | 78 |
| 3 | 2 (3) | CD2Cl2 | 90 | 22 | 0 |
| 4 | 2 (3) | d 8 -THF | 90 | 2.5 | 99 |
| 5 | 2 (3) | d 8 -Toluene | 90 | 2.5 | 92 |
| 6 | 2 (3) | C6D6 | 90 | 1.5 | 99 |
| 7 | 2 (3) | EtOAc | 90 | 3 | 97 |
| 8 | 2 (3) | Anisole | 90 | 9 | 77 |
| 9 | 2 (1.5) | C6D6 | 90 | 37 | 79 |
| 10 | 2 (3) | C6D6 | 50 | 36 | 99 |

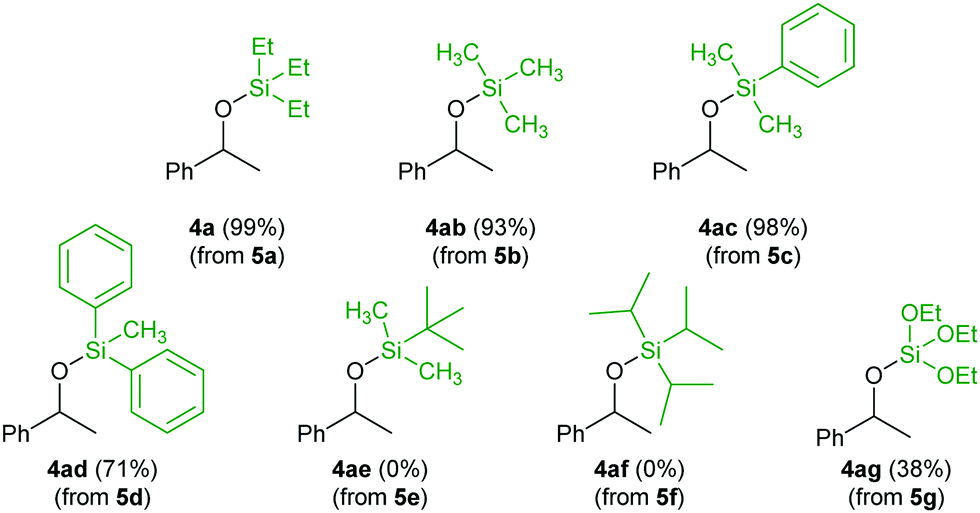
The influence of the silicon coordination sphere on the reactivity was tested by reaction of acetophenone (3a) with different silylformates 5a–g under the optimized conditions (Scheme 2). The reaction worked efficiently with triethyl-, trimethyl- or dimethylphenylsilyl formates (5a–c) and acetophenone (3a), giving compounds 4a–4ac with yields above 93%. It is worthy to highlight that the possibility to use trimethylsilyl formate (5b) represents a major synthetic advantage of the use of these surrogates, because its parent hydrosilane Me3SiH is gaseous. The increase of the bulkiness on the substituents around the silicon core implied a decrease in the yield for the transformation. While methyldiphenylsilylated alcohol 4ad was still obtained in 71% yield, tert-butyldimethylsilyl and triisopropylsilyl formates (5e and 5f) completely suppressed the reduction of the ketone. Finally, the use of the more acidic triethoxysilyl formate (5g) led to a significant drop of the yield providing the silylated alcohol 4ag in 38% yield. This trend highlights the importance of the steric and electronic parameters of the silyl moiety on the outcome of the reaction.
 | ||
| Scheme 2 Silyl formate scope for the hydrosilylation of acetophenone. 0.1 mmol scale. Yields are determined by 1H NMR with mesitylene as an internal standard. See ESI† for more details. | ||
A number of ketones were thereafter tested for transfer hydrosilylation with triethylsilyl or trimethylsilyl formates (5a and 5b) as hydrosilane surrogates (Scheme 3). Several substituted acetophenones were successfully hydrosilylated in short reaction times. Electron-donating substituents (4b and c) or electron-withdrawing groups (4d–h) were well tolerated with yields above 82%. Remarkably, 4-iodoacetophenone (3e) reacted without any loss of the iodine core. With more challenging ortho substituted acetophenones, 4i and 4j were obtained in 88% and 99% yields, respectively. Elongating the alkyl chain (4k) did not affect the reactivity. However, when phenyl isopropyl ketone (3l) was submitted to the reaction, the yield of hydrosilylated alcohol 4la dropped to 33% due to the higher steric hindrance present in the molecule. Hydrosilylation of this type of substrate could be carried out with higher yield if the less hindered trimethylsilyl formate (5b) was used, providing 4lb in 75% yield. This proves the importance of the steric hindrance for this transformation. Another proof for the importance of this effect was obtained with 4,4′-dimethylbenzophenone (3m). In this case, the reaction with triethylsilyl formate (5a) gave silyl ether 4ma in 89% yield, but required a longer reaction time (42 h). Reducing the bulkiness on the reagent by using trimethylsilyl formate (5b) afforded 4mb with a comparable yield of 76% with a significantly reduced reaction time (13 h). Benzophenone derivatives 3n and 3o were also hydrosilylated in 79% and 99% yields with silyl formate 5a, respectively. In these cases, to perform the transformation within a reasonable reaction time, the amount of silylformate reagent was increased to two equivalents.
 | ||
| Scheme 3 Substrate scope for the transfer hydrosilylation of ketones (0.1 mmol scale). Yields were determined by 1H NMR with mesitylene as an internal standard. Scaled-up reactions (0.5 mmol scale) were performed with toluene as the solvent. Yields of isolated products from scaled-up reactions are given within parentheses. [a] 2 equivalents of 5a were used. [b] Reaction performed in anisole as the solvent. [c] Reaction performed at 60 °C. | ||
Remarkably, compound 4oc bearing a useful dimethylphenylsilyl protecting group was obtained in a 91% yield within 4 h in anisole as the solvent. The reaction proved to be scalable to 0.5 mmol, yielding product 4oc in 63% isolated yield. More challenging substrates, such as trifluoromethylketone 3p and α,β-unsaturated ketones 3q–s,17 were successfully hydrosilylated in 61–99% yields, with a 1,2-selectivity for the latter. Among them, compound 4r was obtained in only 61% yield due to the formation of the conjugated enolether by-product. Heteroaromatic silylated alcohols 4t and 4u were obtained in 81% and 93% yield, respectively. Finally, dialkyl ketones 3v and 3w could also react under these conditions giving a 95% yield of the hydrosilylated products in both cases. Although free alcohols, carboxylic acids, amides or amines did not shut down the reaction, they exhibited a detrimental effect on the yields (see competition reactions in the Table S4, ESI†). The selectivity between ketones and aldehydes was studied in the transfer hydrosilylation of 4-acetylbenzaldehyde (3x) with only one equivalent of silyl formate 5a. Not surprisingly, the aldehyde group was fully hydrosilylated after 2 h of reaction, while the ketone moiety remained intact (Scheme 4A).
 | ||
| Scheme 4 Yields were determined by 1H NMR with mesitylene as an internal standard. (A) Selectivity of the PNHP-based ruthenium catalyst 2 for the transfer hydrosilylation of carbonyl groups (0.1 mmol scale). (B) Deuterosilylation of ketones (0.1 mmol scale). | ||
To verify the origin of the hydride, deuterated silyl formate 5a-d1 was synthesized and submitted to reaction. Deuterosilylated product 4a-d1 was obtained as the only product, confirming that the hydride source is indeed the formate group (Scheme 4B). In addition, the absence of the unlabeled product 4a suggests that the N–H bond on the catalyst ligand is not cleaved during the catalysis.
To evaluate the importance of the role of the N–H bond present in the PNHP ligand on catalyst 2, an analogous complex, where the N–H bond is methylated (2-Me), was synthesized. While catalyst 2 was able to reduce acetophenone (3a) and benzaldehyde (6), the parent 2-Me catalyst could reduce aldehyde 6 but not ketone 3a (Table 2). This observation is consistent with the requirement of the N–H motif for the reduction of ketones.



Based on these observations, a putative mechanism for this transformation is illustrated in Scheme 5. As we previously reported, an initial decarbonylation of silyl formate 5 on catalyst 2 generates the active catalyst ruthenium formate A, which through decarboxylation leads to the ruthenium hydride species B.18 The presence of a ruthenium hydride species was confirmed by NMR analysis of the reaction mixture (see Fig. S8 and S9, ESI†).
 | ||
| Scheme 5 Putative mechanism for the transfer hydrosilylation of ketones with silyl formates. | ||
Interaction of ketone 3 with the ruthenium–hydride complex B results in its reduction, presumably assisted by a hydrogen bond formed between the carbonyl group and the ligand PNHP (C).19 The same type of interaction in the generated intermediate D favours the attack of the alkoxide on the silicon center of a new molecule of silyl formate 5, generating the final hydrosilylated product 4, regenerating the active catalyst species A, and closing the catalytic cycle.
In summary, we have unlocked the possibility of using silyl formates in the transfer hydrosilylation of ketones by selecting a suitable PNHP-based ruthenium catalyst 2. In addition, as shown in the control experiments, evidence of the crucial role of the N–H bond in the catalyst ligand was provided. This transformation opens the possibility of applying silyl formates as hydrosilane surrogates to reduce the more challenging ketones.
Conflicts of interest
There are no conflicts to declare.Notes and references
- (a) B. Marciniec, in Hydrosilylation: A Comprehensive Review on Recent Advances, ed. B. Marciniec, Springer Netherlands, Dordrecht, 2009, pp. 3–51 CrossRef; (b) M. C. Lipke, A. L. Liberman-Martin and T. D. Tilley, Angew. Chem., Int. Ed., 2017, 56, 2260–2294 ( Angew. Chem. , 2017 , 129 , 2298 ) CrossRef CAS PubMed.
- M. Oestreich, Angew. Chem., Int. Ed., 2016, 55, 494 ( Angew. Chem. , 2016 , 128 , 504 ) CrossRef CAS PubMed.
- (a) S. Amrein and A. Studer, Helv. Chim. Acta, 2002, 85, 3559 CrossRef CAS; (b) S. Amrein, A. Timmermann and A. Studer, Org. Lett., 2001, 3, 2357 CrossRef CAS PubMed; (c) A. Studer and S. Amrein, 39 , Angew. Chem., Int. Ed., 2000, 3080 ( Angew. Chem. , 2016 , 112 , 3196 ) CrossRef CAS.
- (a) A. Simonneau and M. Oestreich, Angew. Chem., Int. Ed., 2013, 52, 11905 ( Angew. Chem. , 2013 , 125 , 12121 ) CrossRef CAS PubMed; (b) S. Keess, A. Simonneau and M. Oestreich, Organometallics, 2015, 34, 790 CrossRef CAS.
- C. Chauvier, P. Thuéry and T. Cantat, Angew. Chem., Int. Ed., 2016, 55, 14096 ( Angew. Chem. , 2016 , 128 , 14302 ) CrossRef CAS PubMed.
- (a) P. K. Sahoo, T. Zhang and S. Das, Eur. J. Org. Chem., 2021, 1331 CrossRef CAS; (b) D. Bulushev and J. R.-H. Ross, ChemSusChem, 2018, 11, 821 CrossRef CAS PubMed.
- (a) W. Leitner, Angew. Chem., Int. Ed. Engl., 1995, 34, 2207 ( Angew. Chem. , 1995 , 107 , 2391 ) CrossRef CAS; (b) S. Moret, P. J. Dyson and G. Laurenzcy, Nat. Commun., 2014, 5, 1 Search PubMed.
- T. Godou, C. Chauvier, P. Thuéry and T. Cantat, Synlett, 2017, 2473 CAS.
- C. Chauvier, T. Godou and T. Cantat, Chem. Commun., 2017, 53, 11697 RSC.
- (a) P. A. Dub, B. L. Scott and J. C. Gordon, J. Am. Chem. Soc., 2017, 139, 1245 CrossRef CAS PubMed; (b) S. Werkmeister, K. Junge, B. Wendt, E. Alberico, H. Jiao, W. Baumann, H. Junge, F. Gallou and M. Beller, Angew. Chem., Int. Ed., 2014, 53, 8722 ( Angew. Chem. , 2014 , 126 , 8867 ) CrossRef CAS PubMed.
- (a) T. Zell and D. Milstein, Acc. Chem. Res., 2015, 48, 1979 CrossRef CAS PubMed; (b) J. Zhang, G. Leitus, Y. Ben-David and D. Milstein, Angew. Chem., Int. Ed., 2006, 45, 1113 ( Angew. Chem. , 2006 , 118 , 1131 ) CrossRef CAS PubMed; (c) T. Zell, Y. Ben-David and D. Milstein, Angew. Chem., Int. Ed., 2014, 53, 4685 ( Angew. Chem. , 2014 , 126 , 4773 ) CrossRef CAS PubMed; (d) J. O. Bauer, S. Chakraborty and D. Milstein, ACS Catal., 2017, 7, 4462 CrossRef CAS.
- V. Papa, J. R. Cabrero-Antonino, E. Alberico, A. Spanneberg, K. Junge, H. Junge and M. Beller, Chem. Sci., 2017, 8, 3576 RSC.
- (a) D. Spasyuk, C. Vicent and D. G. Gusev, J. Am. Chem. Soc., 2015, 137, 3743 CrossRef CAS PubMed; (b) D. G. Gusev, ACS Catal., 2016, 6, 6967 CrossRef CAS.
- W. Kuriyama, T. Matsumoto, O. Ogata, Y. Ino, K. Aoki, S. Tanaka, K. Ishida, T. Kobayashi, N. Sayo and T. Saito, Org. Process Res. Dev., 2012, 16, 166 CrossRef CAS.
- (a) S. Chakraborty, H. Dai, P. Bhattacharya, N. T. Fairweather, M. S. Gibson, J. A. Krause and H. Guan, J. Am. Chem. Soc., 2014, 136, 7869 CrossRef CAS PubMed; (b) T. Otsuka, A. Ishii, P. A. Dub and T. Ikariya, J. Am. Chem. Soc., 2013, 135, 9600 CrossRef CAS PubMed; (c) S. Gao, W. Tang, M. Zhang, C. Wang and J. Xiao, Synlett, 2016, 1748 CAS; (d) X. Han, L. Rong, J. Wu, L. Zhang, Z. Wang and K. Ding, Angew. Chem., Int. Ed., 2012, 51, 13041 ( Angew. Chem. , 2012 , 124 , 13218 ) CrossRef PubMed; (e) L. A. Suàrez, Z. Culakova, D. Balcells, W. Bernskoetter, O. Eisenstein, K. I. Goldberg, N. Hazari, M. Tilset and A. Nova, ACS Catal., 2018, 8, 8751 CrossRef.
- (a) D. Peng, Y. Zhang, X. Du, L. Zhang, X. Leng, M. D. Walter and Z. Huang, J. Am. Chem. Soc., 2013, 135, 19154 CrossRef CAS PubMed; (b) M. L. Scheuermann, S. P. Semproni, I. Pappas and P. J. Chirik, Inorg. Chem., 2014, 53, 9463 CrossRef CAS PubMed.
- (a) J. Wang, R. Qin, H. Fu, J. Chen, J. Feng, H. Chen and X. Li, Tetrahedron: Asymmetry, 2007, 18, 847 CrossRef CAS; (b) G. Z. Zheng and T. H. Chan, Organometallics, 1995, 14, 70 CrossRef CAS; (c) Y. Sumida, H. Yorimitsu and K. Oshima, J. Org. Chem., 2009, 74, 7986 CrossRef CAS PubMed; (d) N. Ikeda and T. Konno, J. Fluorine Chem., 2015, 173, 69 CrossRef CAS; (e) M. Rubio, J. Campos and E. Carmona, Org. Lett., 2011, 13, 5236 CrossRef CAS PubMed.
- C. Chauvier, A. Imberdis, P. Thuéry and T. Cantat, Angew. Chem., Int. Ed., 2020, 132, 14123 ( Angew. Chem. , 2020 , 132 , 14123 ) CrossRef.
- (a) A. Kaithal, M. Schmitz, M. Hölscher and W. Leitner, ChemCatChem, 2020, 12, 781 CrossRef CAS; (b) A. Passera and A. Mezzetti, Adv. Synth. Catal., 2019, 361, 4691 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2cc00666a |
| This journal is © The Royal Society of Chemistry 2022 |