
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Highly stable silver nanohybrid electrocatalysts for the oxygen reduction reaction†
Quentin
Lenne
a,
Maurice
Retout
 b,
Bryan
Gosselin
b,
Gilles
Bruylants
b,
Ivan
Jabin
c,
Jonathan
Hamon
d,
Corinne
Lagrost
*a and
Yann R.
Leroux
*a
b,
Bryan
Gosselin
b,
Gilles
Bruylants
b,
Ivan
Jabin
c,
Jonathan
Hamon
d,
Corinne
Lagrost
*a and
Yann R.
Leroux
*a
aUniv. Rennes, CNRS, ISCR – UMR 6226, 35000 Rennes, France. E-mail: yann.leroux@univ-rennes1.fr
bEMNS, Université libre de Bruxelles (ULB), avenue F. D. Roosevelt 50, CP165/64, B-1050 Brussels, Belgium
cLCO, Université libre de Bruxelles (ULB), CP 160/06, avenue F. D. Roosevelt 50, 1050 Brussels, Belgium
dIMN, 2 rue de la Houssinière, 44000 Nantes, France
First published on 16th February 2022
Abstract
Silver nanoparticles (AgNPs) were deliberately functionalized via aryl diazonium chemistry with a monolayer of calix[4]arenes. The resulting nanohybrids show high efficiency and high selectivity toward the ORR in alkaline media along with an exceptional durability and a high methanol tolerance.
Catalysts for the oxygen reduction reaction (ORR) are key components for implementing renewable-energy technologies. The slow kinetics of the ORR is still a major bottleneck and the reaction requires not only efficient and durable but also affordable electrocatalysts. Platinum1 and platinum-based2,3 materials are known to be the best ORR electrocatalysts in acidic media but they suffer from high cost and low catalytic stability. Moreover, highly reactive oxygen species (ROS) produced during the ORR were shown to lead to the deactivation or even the dissolution of the catalyst during operation.4–8 This has driven many research groups to look to alternative and cheaper catalysts. In this context, interest has been focused on silver materials. They are good ORR electrocatalysts in alkaline media at a much lower cost, although still much less studied than gold structures.9 Apart from stability, a key issue for ORR activity is whether the reduction of O2 proceeds by two electrons to H2O2 (or HO2−) or by four electrons to H2O (or HO−). The last pathway is the most preferred one for energy applications due to the production of higher currents and limitation of H2O2 decomposition side reactions, possibly harmful for the fuel cell components. Several decades ago, the ability of bulk silver (Ag) to reduce oxygen in alkaline aqueous solutions through a 4-electron or 2-electron pathways was demonstrated, depending on the electrode potential.10 Silver nanomaterials were then successfully employed for the ORR, with various nanostructures including nanodecahedra,11 triangular nanoplates,12 nanowires13 or nanocubes.14 The size effect of silver nanoparticles (AgNPs) was much less studied than that of gold nanoparticles but, as a general trend, 4-electron pathway was found to be favoured on large AgNPs whereas 2-electron pathway prevailed on small ones at low overpotentials.15,16 However, nanoclusters (2–5 silver atoms) were also demonstrated to efficiently catalyse the 4-electron reduction of O2,17 but in contrast to earlier work.18
Far to have an adverse impact on the catalytic performances, the deliberate surface functionalization of metallic nanoparticles with organic ligands has been recently demonstrated to be an emerging strategy for enhancing selectivity and/or stability in electrocatalysis.19–22 Yet, such a strategy has been mainly applied to gold or platinum nanomaterials for the ORR, and hardly explored with silver nanoparticles.19 In addition, the nature of the interaction between the nanoparticles and the ligands along with their organisation with respect to the surface need to be further considered to fully unveil the potential of this strategy.
Herein, silver nanoparticles (AgNPs) of 6 nm and 18 nm functionalized by a covalently-bound thin layer of calix[4]arenes bearing carboxylate groups (calix-AgNPs, Scheme 1) are evaluated as nanocatalysts for the ORR.23 The rigid macrocyclic structure of the calixarene platform ensures a strong anchoring of the organic moiety through multiple anchoring points and allows a very good spatial organization of the interface.24 Furthermore, the carboxylate groups warrant high stability and dispersity of the synthesized nanoparticles in alkaline aqueous medium. The ORR activity of these calix-AgNPs is compared to that of classical 5 nm and 20 nm citrate-stabilized AgNPs (citrate-AgNPs).
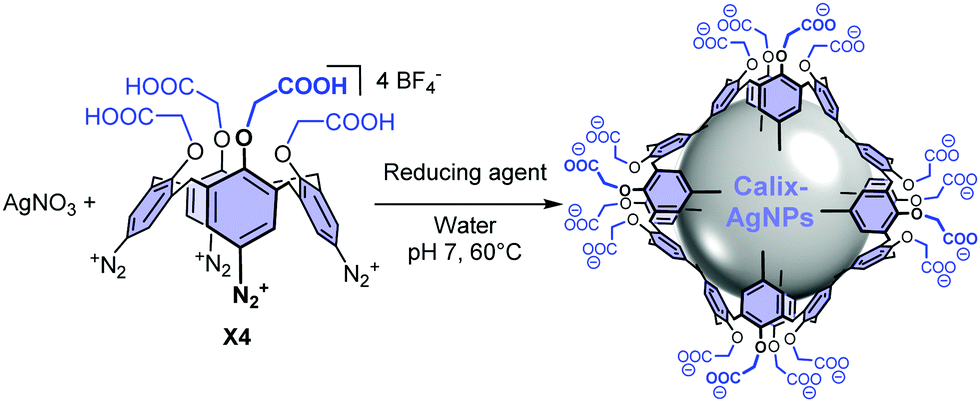 | ||
| Scheme 1 Synthesis of silver nanoparticles functionalized by a thin layer of calix[4]arenes bearing carboxylate arms (calix-AgNPs). | ||
Calix-AgNPs were synthesized in a one-pot procedure that consists in the simultaneous reduction of AgNO3 and of calix[4]arene-tetraacetic acid-tetradiazonium salt X4 at pH7 (Scheme 1).23 The size of the resulting NPs can be tuned through the choice of the reducing agent, NaBH4 and sodium ascorbate, leading to 6 and 18 nm NPs, respectively. The calix-AgNPs were fully characterized by UV-vis absorption and IR transmission spectroscopies, TEM, and X-ray photoelectrons spectroscopy (see ESI†). Commercially available citrate-AgNPs of 5 nm and 20 nm are used for comparison. As shown previously, the citrate-based ligands are merely physisorbed onto the NPs surface and form disordered multilayers.25 The surface chemistry of the two series of AgNPs was investigated by XPS (Fig. S3 and S4, ESI†), demonstrating the presence of the organic ligands onto metallic silver surfaces in all cases (see ESI†).
The electrocatalytic activities of the different AgNPs were measured using a platinum (Pt) ring/glassy carbon (GC) disk rotating ring-disk electrode (RRDE) in O2-saturated aqueous 0.1 M KOH solution. The AgNPs were deposited onto the GC disk surface with the same mass loading (283 μg cm−2). This value was chosen to maximize the loading on the carbon disk electrode in order to limit the contribution of the underlying carbon surface itself. It is well-known that carbon surfaces promote a two-electron reduction of O2 in alkaline media, then substrate issues may complicate the determination of the number of exchanged electrons. Fig. 1 presents the results obtained with the smallest calix-AgNPs and citrate-AgNPS, i.e. 6 and 5 nm, respectively. The onset potentials (i.e. potentials corresponding to 5% of the diffusion limiting current) were found equal to 0.74 V and 0.71 V (vs. RHE) for calix-AgNPs and citrate-AgNPs respectively, while half-wave potentials were determined as 0.50 V and 0.47 V (vs. RHE) respectively. Such values are in line with those reported for silver-based catalysts under similar conditions (0.1 M KOH) (see Table S1, ESI†).18,26,27 The yield of hydroperoxide ions (HO2−) generated during ORR was monitored by collection experiments at the Pt ring electrode (Fig. 1, top curves). The Pt ring was set to a potential of +1.70 V (vs. RHE) to promote HO2− oxidation. Almost no HO2− was generated with the calix-AgNPs catalysts during the ORR whereas it was significantly detected for citrate-AgNPs.
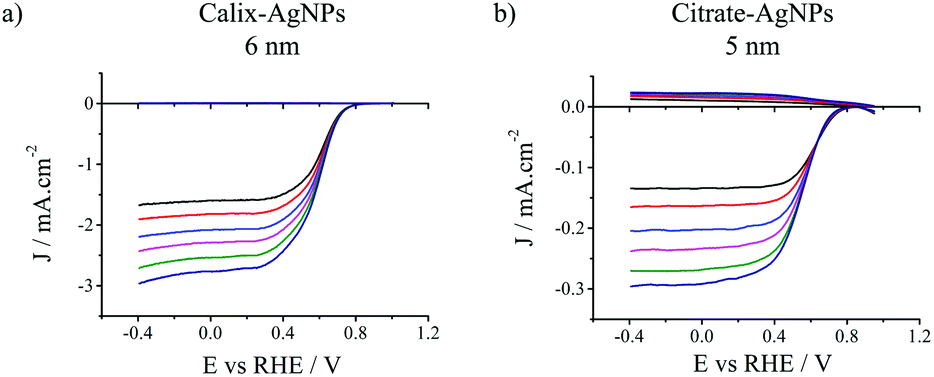 | ||
| Fig. 1 RRDE voltammetric curves in O2-saturated 0.1 M KOH solution of GC disk electrodes modified with either (a) 6 nm calix-AgNPs or (b) 5 nm citrate-AgNPs (bottom curves). Ring electrode (top curves): Pt. v = 10 mV s−1. ω = 400, 600, 900, 1200, 1600, 2000 rpm. Ering = 1.70 V vs. RHE. Normalized current density per electrochemically active surface area are reported. | ||
Data processing was performed on RRDE experiments to follow the yield of HO2− and the number of electrons n exchanged during the ORR process. The number of electrons exchanged can be quantified using the disk and ring currents according to:
 | (1) |
 | (2) |
Fig. 2 displays the variation of n (black lines) and % HO2− (blue lines) as a function of the potential during the ORR for both calix-AgNPs (6 nm) and citrate-AgNPs (5 nm). A constant n value of ca. 4 over the entire potential range is obtained for calix-AgNPs together with a very low production of HO2− (<1%). Such a behaviour evidences an efficient 4-electron process. Citrate-AgNPs also display n value close to 4, between 3.6 and 3.7, but with higher production level of HO2− (up to 18%). This suggests that the oxygen reduction proceeds simultaneously through a 2-electron and a 4-electron processes. Similar experiments were performed with calix-AgNPs and citrate-AgNPs of 18 and 20 nm respectively (Fig. S5 and S6, ESI†). Onset potentials of 0.71 V and 0.72 V (vs. RHE) were measured for calix-AgNPs and citrate-AgNPs respectively, while half-wave potentials were found to be 0.49 V and 0.48 V (vs. RHE), respectively. For 18 nm calix-AgNPs, n values of 3.8–3.9 over the entire potential range was calculated with HO2− production level around 6–8%, whereas n value of 4 and HO2− production level below 1% were measured for 20 nm citrate-AgNPs.
 | ||
| Fig. 2 Potential dependence of the number of electrons n (black lines) and the yield of HO2− (blue lines) produced during ORR experiments for (a) 6 nm calix-AgNPs and (b) 5 nm citrate-AgNPs. | ||
At this stage, it is thus interesting to consider the ORR selectivity regarding the size of the AgNPs since it has been earlier demonstrated to impact the 2 or 4-electron pathways. Here, using citrate-stabilized AgNPs, 4-electron pathway is favoured on the larger 20 nm citrate-AgNPs whereas on the smaller 5 nm citrate-AgNPs simultaneous 2-electron and a 4-electron processes are observed. This is in line with a more favourable ORR process to H2O (OH−) on larger AgNPs, as described in the literature.15,16 Interestingly, the results obtained with calix-AgNPs show an opposite trend. The performances of calix-AgNPs of 6 and 18 nm are rather close yet significantly better with the smallest NPs, notably considering the lower yield of HO2− that is produced (Table 1). It is further interesting to compare the electrocatalytic activities of the two series of AgNPs. Focusing on the smallest AgNPs, the calix-AgNPs outperform citrate-AgNPs, with higher onset or half-wave potentials and better selectivity towards the 4-electron pathway, whereas an opposite trend is observed on the bigger AgNPs. Finally, the 6 nm calix-AgNPs and the 20 nm citrate-AgNPs have similar electrocatalytic activities. In order to report the catalytic efficiency of our new nanohybrids, we calculate the normalized kinetic current density per electrochemically active surface area (ECSA, ESI†) at 0.6 V vs. RHE (Table 1). The surface activities of calix-AgNPs were found to be significantly larger than those of citrate-AgNPs. The smallest calix-AgNPs even led to the highest activity. Note that the ECSA of calix-AgNPs is smaller than the one of citrate-AgNPs (Table S1, ESI†). It can be explained by the covalent binding of the calix[4]arenes to the AgNPs surface that may potentially decrease the number of available catalytic sites, whereas citrate molecules are simply physisorbed onto the particles. The mass-transfer corrected Tafel plots lead to slopes of ca. 80 mV per decade at low overpotentials (low J) for all the nanocatalysts (Fig. S7, ESI†). These values are close to the typical slope of 60 mV per decade generally found for the ORR and agree with the values reported in the literature,18,28–30 suggesting that the rate determining step is the first electron transfer as reported for platinum (Pt) and gold (Au). The modification of the electrocatalytic properties of the AgNPs upon functionalization with calix[4]arenes monolayer may be ascribed to a ligand effect, strengthened by the robust grafting of the organic ligand onto the surface. Recent studies have shown that the electronic interactions between metal nanoparticles and ligands play a significant role in determining the electrocatalytic activity as a result of the manipulation of the electronic energy of the metal nanoparticles and hence the interactions with oxygen.31–33 While DFT calculations of elementary step energetics could provide valuable information,31 it is out of the scope of this communication.
| 5 nm citrate-AgNPS | 20 nm citrate-AgNPs | 6 nm calix-AgNPs | 20 nm calix-AgNPs | |
|---|---|---|---|---|
| a The number of electron n is given for the entire potential range, i.e. from 0.6 to −0.4 V vs. RHE. | ||||
| n | 3.6–3.7 | 4 | 4 | 3.8–3.9 |
| % HO2− | 14–18% | <1% | <1% | 6–8% |
| Onset potential (vs. RHE) | 0.71 V | 0.72 V | 0.74 V | 0.71 V |
| Half-wave potential (vs. RHE) | 0.47 V | 0.48 V | 0.50 V | 0.49 V |
| Surface activity @ 0.6 V vs. RHE | 0.14 mA cm−2 | 0.26 mA cm−2 | 1.95 mA cm−2 | 0.79 mA cm−2 |
| % Loss of surface activity @ 0.6 V vs. RHE after stability test | 55% | 35% | 3% | 22% |
One crucial point for electrocatalysts concerns their durability, which is of paramount importance in applications such as fuel cells. The long-term stability of the two series of AgNPs was evaluated through chronoamperometric experiments at a constant voltage of 0.55 V (vs. RHE) for 12 h in 0.1 M KOH O2-saturated solution (Fig. S8, ESI†). Fig. 3 presents LSV curves at 1600 rpm of calix-AgNPs (6 nm) and citrate-AgNPs (5 nm) before and after the 12 h-chronoamperometry. The corresponding surface activities were deduced from the recorded LSV. A loss of only 3% in the surface activity of 6 nm calix-AgNPs is observed whereas a loss up to 55% is measured for 5 nm citrate-AgNPs. Similarly, 22% and 35% losses in surface activity are observed on the 18 nm calix-AgNPs and 20 nm citrate-AgNPs, respectively (Table 1, Fig. S9, ESI†). The results indicate that the covalently bound calix[4]arene monolayers dramatically enhance the stability of the silver nanocatalysts in contrast to the physisorbed and loosely packed citrates.
 | ||
| Fig. 3 Linear sweep voltammetry (LSV) curves recorded at 1600 rpm on (a) 6 nm calix-AgNPs and (b) 5 nm citrates AgNPs before (black curves) and after (red curves) stability test in O2-saturated 0.1 M KOH solution. Normalized current density per electrochemically active surface area are reported. | ||
XPS analyses were further conducted before and after the accelerated stability tests, which consist in one thousand cyclic voltammograms at 500 mV s−1 between +1.0 and −0.4 V vs. RHE in O2-saturated 0.1 M KOH solution (Fig. S10–S13, ESI†) The chemical states of the silver nanomaterials were especially scrutinized through the determination of the modified Auger parameter of silver α’ for a more accurate analysis34,35 (see ESI†). The XPS analyses of the silver nanoparticles evidence an oxidation of the silver nanoparticles after the accelerated stability tests when stabilized by citrates. The effect is stronger with the smallest 5 nm citrate-AgNPs. In sharp contrast, the calix-AgNPs retain their metallic character after operation, even for the small 6 nm calix-AgNPs. This may explain the loss in surface activity of the citrate-AgNPs, especially for the 5 nm NPs. Thus, the grafting of calix[4]arenes onto the nanoparticle's surface provides a highly robust but very thin organic interface that strengthens the catalytic stability of the resulting calix-AgNP nanohybrids. Note that the stability obtained with 6 nm calix-AgNPs is by far one of the best compared to those reported in the literature for silver nanomaterials.15,26,36
Finally, we evaluated the methanol tolerance of the calix-AgNPs (6 nm) because this aspect may be also of great importance for ORR catalyst applications, namely direct methanol fuel cells (DMFC). In DMFC, the crossover of methanol through the membrane could be an issue. For instance, Pt nanocatalysts are very sensitive to this crossover which deactivates them. Remarkably, the addition of methanol caused only a very weak current fluctuation, demonstrating the great methanol tolerance of the 6 nm calix-AgNPs (Fig. S14, ESI†).
Herein, we demonstrate that the deliberate functionalization of AgNPs with a covalently-bound thin layer of calix[4]arenes dramatically enhances the ORR performances of silver nanocatalysts in alkaline media compared to AgNPs stabilized with citrates. The calix-AgNPs outperform citrate-AgNPs in terms of efficiency (surface activity) and durability, suggesting that the surface modification procedure has a great impact on the performances of the material. Our own experiments on citrate-AgNPs confirm that the 4-electron pathway is favoured on large citrate-AgNPs whereas the 2-electron pathway prevails on small ones. However, we demonstrate that the deliberate functionalization of AgNPs with a thin layer of calix[4]arenes reverses this trend, allowing an optimum use of silver materials as catalysts in the ORR. In particular, the smallest calix-AgNPs (6 nm) manifest an exceptional durability with only a loss of 3% in mass activity after 12 h ORR operation together with a great tolerance to methanol. This calixarene-based coating strategy opens perspectives to the use of such nanohybrids in fuel cells and notably DMFCs.
We thank Dr Ludovic Troian-Gautier for the synthesis of calix[4]arene X4 and Dr Antoine Vacher for IR spectra. This work was supported by the Comité National de la Recherche Scientifique (CNRS) and the Universities of Rennes 1 and Bruxelles.
Conflicts of interest
M. R. was a postdoctoral researcher for X4C from August 2020 to January 2021. I. J. and C. L. are shareholders of X4C. I. J. and G. B. are consultants for X4C. All other authors declare that they have no conflict of interest.Notes and references
- Y. Wang and P. B. Balbuena, J. Phys. Chem. B, 2005, 109, 18902–18906 CrossRef CAS PubMed.
- J. Wu and H. Yang, Acc. Chem. Res., 2013, 46, 1848–1857 CrossRef CAS PubMed.
- D. Wu, X. Shen, Y. Pan, L. Yao and Z. Peng, ChemNanoMat, 2020, 6, 32–41 CrossRef CAS.
- J.-M. Noël, A. Latus, C. Lagrost, E. Volanschi and P. Hapiot, J. Am. Chem. Soc., 2012, 134, 2835–2841 CrossRef PubMed.
- S. Lhenry, Y. R. Leroux and P. Hapiot, Anal. Chem., 2012, 84, 7518–7524 CrossRef CAS PubMed.
- S. J. Percival, J. E. Dick and A. J. Bard, Anal. Chem., 2017, 89, 3087–3092 CrossRef CAS PubMed.
- J.-M. Noël, Y. Yu and M. V. Mirkin, Langmuir, 2013, 29, 1346–1350 CrossRef PubMed.
- M. Umeda, T. Maruta, M. Inoue and A. Nakazawa, J. Phys. Chem. C, 2008, 112, 18098–18103 CrossRef CAS.
- H. Erikson, A. Sarapuu and K. Tammeveski, ChemElectroChem, 2019, 6, 73–86 CrossRef CAS.
- N. A. Shumilova, G. V. Zhutaeva and M. P. Tarasevich, Electrochim. Acta, 1966, 11, 967–974 CrossRef CAS.
- Q. Wang, X. Cui, W. Guan, L. Zhang, X. Fan, Z. Shi and W. Zheng, J. Power Sources, 2014, 269, 152–157 CrossRef CAS.
- C.-L. Lee, H.-P. Chiou, C.-M. Syu and C.-C. Wu, Electrochem. Commun., 2010, 12, 1609–1613 CrossRef CAS.
- S. M. Alia, K. Duong, T. Liu, K. Jensen and Y. Yan, ChemSusChem, 2012, 5, 1619–1624 CrossRef CAS PubMed.
- C.-L. Lee, Y.-L. Tsai, C.-H. Huang and K.-L. Huang, Electrochem. Commun., 2013, 29, 37–40 CrossRef CAS.
- Y. Yang and Y. Zhou, J. Electroanal. Chem., 1995, 397, 271–278 CrossRef.
- J.-J. Han, N. Li and T.-Y. Zhang, J. Power Sources, 2009, 193, 885–889 CrossRef CAS.
- X. Yang, L. Gan, C. Zhu, B. Lou, L. Han, J. Wang and E. Wang, Chem. Commun., 2014, 50, 234–236 RSC.
- Y. Lu and W. Chen, J. Power Sources, 2012, 197, 107–110 CrossRef CAS.
- Q. Lenne, Y. R. Leroux and C. Lagrost, ChemElectroChem, 2020, 7, 2345–2363 CrossRef CAS.
- Q. Lenne, A. Mattiuzzi, I. Jabin, N. Le Poul, Y. R. Leroux and C. Lagrost, Adv. Mater. Interfaces, 2020, 7, 2001557 CrossRef CAS.
- D.-H. Nam, P. De Luna, A. Rosas-Hernández, A. Thevenon, F. Li, T. Agapie, J. C. Peters, O. Shekhah, M. Eddaoudi and E. H. Sargent, Nat. Mater., 2020, 19, 266–276 CrossRef CAS PubMed.
- P. Hu, L. Chen, X. Kang and S. Chen, Acc. Chem. Res., 2016, 49, 2251–2260 CrossRef CAS PubMed.
- M. Retout, I. Jabin and G. Bruylants, ACS Omega, 2021, 6, 19675–19684 CrossRef CAS PubMed.
- L. Troian-Gautier, A. Mattiuzzi, O. Reinaud, C. Lagrost and I. Jabin, Org. Biomol. Chem., 2020, 18, 3624–3637 RSC.
- J.-W. Park and J. S. Shumaker-Parry, J. Am. Chem. Soc., 2014, 136, 1907–1921 CrossRef CAS PubMed.
- A. Treshchalov, H. Erikson, L. Puust, S. Tsarenko, R. Saar, A. Vanetsev, K. Tammeveski and I. Sildos, J. Colloid Interface Sci., 2017, 491, 358–366 CrossRef CAS PubMed.
- Y. Lu, Y. Wang and W. Chen, J. Power Sources, 2011, 196, 3033–3038 CrossRef CAS.
- B. B. Blizanac, P. N. Ross and N. M. Marković, J. Phys. Chem. B, 2006, 110, 4735–4741 CrossRef CAS PubMed.
- M. Chatenet, L. Genies-Bultel, M. Aurousseau, R. Durand and F. Andolfatto, J. Appl. Electrochem., 2002, 32, 1131–1140 CrossRef CAS.
- L. Tammeveski, H. Erikson, A. Sarapuu, J. Kozlova, P. Ritslaid, V. Sammelselg and K. Tammeveski, Electrochem. Commun., 2012, 20, 15–18 CrossRef CAS.
- A. Holewinski, J.-C. Idrobo and S. Linic, Nat. Chem., 2014, 6, 828–834 CrossRef CAS PubMed.
- S.-A. Park, H. Lim and Y.-T. Kim, ACS Catal., 2015, 5, 3995–4002 CrossRef CAS.
- W. Seh Zhi, J. Kibsgaard, F. Dickens Colin, I. Chorkendorff, K. Nørskov Jens and F. Jaramillo Thomas, Science, 2017, 355, eaad4998 CrossRef PubMed.
- S. Bera, P. Gangopadhyay, K. G. M. Nair, B. K. Panigrahi and S. V. Narasimhan, J. Electron Spectrosc. Relat. Phenom., 2006, 152, 91–95 CrossRef CAS.
- S. Zanna, C. Saulou, M. Mercier-Bonin, B. Despax, P. Raynaud, A. Seyeux and P. Marcus, Appl. Surf. Sci., 2010, 256, 6499–6505 CrossRef CAS.
- R. Zhou and S. Z. Qiao, Chem. Mater., 2014, 26, 5868–5873 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d2cc00637e |
| This journal is © The Royal Society of Chemistry 2022 |