
On-surface synthesis of a phenylene analogue of nonacene†
Irena
Izydorczyk
 a,
Otilia
Stoica
bc,
Mariusz
Krawiec
*d,
Rémi
Blieck
b,
Rafal
Zuzak
a,
Marcin
Stępień
e,
Antonio M.
Echavarren
*bc and
Szymon
Godlewski
*a
a,
Otilia
Stoica
bc,
Mariusz
Krawiec
*d,
Rémi
Blieck
b,
Rafal
Zuzak
a,
Marcin
Stępień
e,
Antonio M.
Echavarren
*bc and
Szymon
Godlewski
*a
aCentre for Nanometer-Scale Science and Advanced Materials, NANOSAM, Faculty of Physics, Astronomy and Applied Computer Science, Jagiellonian University, Łojasiewicza 11, PL 30-348 Krakow, Poland. E-mail: szymon.godlewski@uj.edu.pl
bInstitute of Chemical Research of Catalonia (ICIQ), Barcelona Institute of Science and Technology, Avenida Països Catalans 16, 43007 Tarragona, Spain. E-mail: aechavarren@iciq.es
cDepartament de Química Organica i Analítica, Universitat Rovira i Virgili, C/Marcell·lí Domingo s/n, 43007 Tarragona, Spain
dInstitute of Physics, Maria Curie-Sklodowska University, Pl. M. Curie-Skłodowskiej 1, 20-031 Lublin, Poland. E-mail: mariusz.krawiec@umcs.pl
eWydział Chemii, Uniwersytet Wrocławski, ul. F. Joliot-Curie 14, 50-383 Wrocław, Poland
First published on 1st March 2022
Abstract
Cyclobuta[1,2-b:3,4-b′]ditetracene – an analogue of nonacene with a cyclobutadiene unit embedded in the central part has been synthesized by the combination of solution and on-surface chemistry. The atomic structure and electronic properties of the product on Au(111) have been determined by high resolution scanning tunnelling microscopy/spectroscopy corroborated by density functional theory calculations. Structural and magnetic parameters derived from theoretical calculations reveal that π conjugation is dominated by radialene-type contribution, with an admixture of cyclobutadiene-like antiaromaticity.
Acenes are a class of polycyclic aromatic hydrocarbons (PAHs) composed of n-linearly fused benzene rings, with promising applications as semiconductors in molecular electronics, due to their distinctive properties, such as narrow HOMO–LUMO gaps.1 However, the instability which grows with the increasing acene length poses a limitation for their use in molecular devices. One of the strategies to overcome this issue is the integration of 4π-electron antiaromatic cyclobutadiene rings in the acene core, increasing the number of aromatic sextets, while keeping their linear shape.2 This approach was first introduced by Vollhardt in the synthesis of [N]-phenylenes,3 a family of conjugated ladder structures with antiaromatic character. Later on, Swager,2a Xia4 and Miao5 extended this concept to the preparation of several phenylene-containing oligoacenes (POAs).2 The latter examples were then confirmed as effective p-type semiconductors in transistor devices, due to their favourable π–π stacking in the crystal arrangement and high charge carrier mobility.4b,5 In recent years surface-assisted synthesis has become a reliable method for the synthesis of cyclobutadiene-containing acenes delivering these intriguing molecules by formal [2+2]-cycloadditions.6
Here we report the straightforward synthesis and scanning tunnelling microscopy/spectroscopy (STM/STS) characterization of the cyclobuta[1,2-b:3,4-b′]ditetracene (1, CBDT), an antiaromatic analogue of nonacene7 containing a 4-membered ring in the central part of the molecule. The on-surface synthesis of compound 1 (CBDT) has been achieved by thermally triggered combination of the intramolecular Ullmann-like coupling,8 together with the dehydrogenation of methylene units present in the starting material 2a (Scheme 1).7 Precursor 2a was readily prepared from known 2,2′-dibromo-4,4′-diiodo-1,1′-biphenyl (3)9 by double Sonogashira coupling with 1,7-enyne 410 to give 5, followed by a double gold(I)-catalysed [4+2] cycloaddition,7,10 to give 2a, together with its regioisomer 2b in a 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio (Scheme 2).
:1 ratio (Scheme 2).
 | ||
| Scheme 1 On-surface synthesis of cyclobuta[1,2-b:3,4-b′]ditetracene (1, CBDT) by a combination of cyclisation and dehydrogenation of 2a. | ||
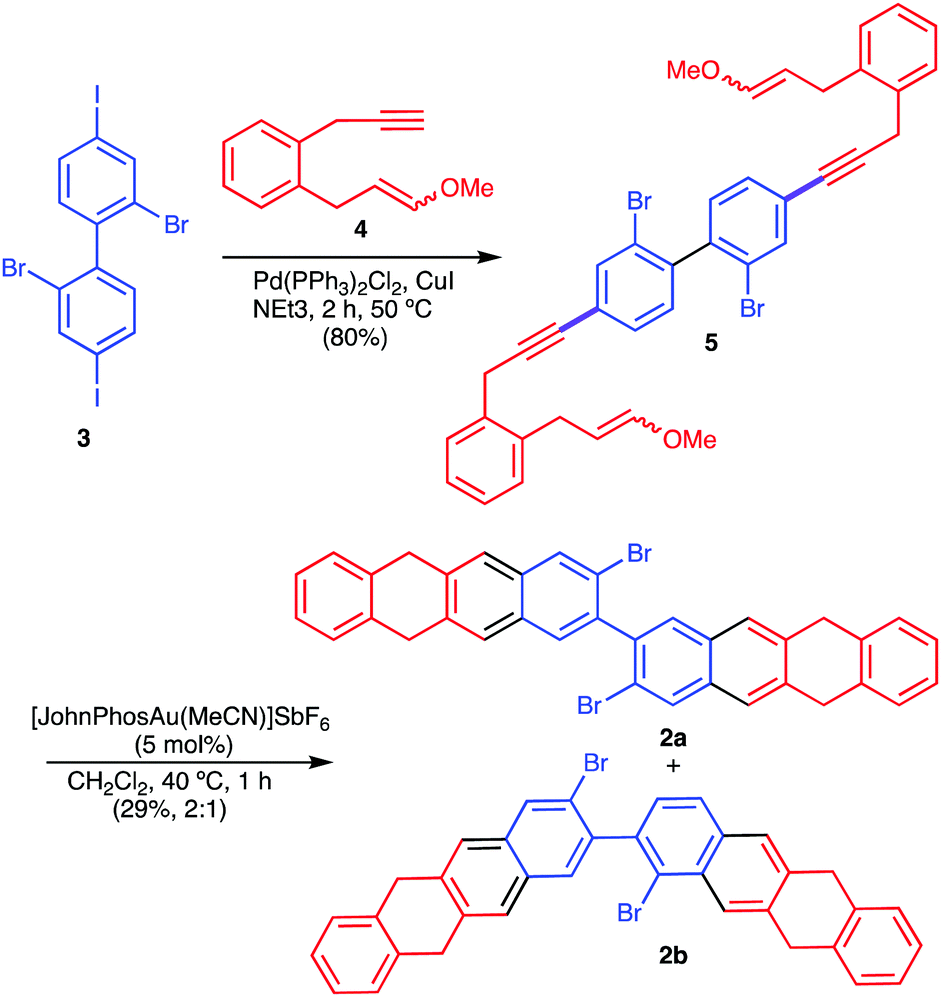 | ||
| Scheme 2 Synthesis of 2a by double gold(I)-catalysed [4+2] cycloaddition of bis 1,7-enyne 5. | ||
After deposition of 2a/2b on Au(111) at 490 K and annealing at 610 K for 15 min, the resulting products are found separated and preferentially located either in the vicinity of the surface herringbone pattern elbows or over the fcc surface region as shown in the STM image (Fig. 1a).7,11 Close inspection of the STM topography led to the conclusion that four differently shaped molecular species could be discerned, which for clarity are marked by dashed red, violet, green or yellow rectangles corresponding to two isomers of cyclobutaditetracene (red and violet rectangles, Fig. 1a and b) or the dimers of tetracene units (yellow and green rectangles, Fig. 1a and b).
 | ||
| Fig. 1 (a) Filled state STM topographic image of the molecules generated through surface-assisted reactions on Au(111), the dashed coloured rectangles mark four different identified moieties, i.e.1 (CBDT, red), 1′ (violet) and tetracene dimers 6, 7 (green and yellow); (b) structural models of the molecules identified in panel (a); (c) and (d) high resolution STM and BR-STM images of CBDT; scale bar: 0.5 nm (e) optimized structural model of 1 (CBDT) adsorbed on Au(111); scanning parameters: (a) 1 V, 25 pA, (c) 10 mV, 30 pA, (d) 5 mV. | ||
The target compound 1 (CBDT) is generated by the cyclisation through the Ullmann-type coupling between the two tetracene moieties equipped with Br atoms. A similar transformation had been demonstrated for the on-surface synthesis of the dibenzo[b,h]biphenylene on Ag(111).8d The cyclisation of 2a/2b takes place with concomitant aromative dehydrogenation of four methylene groups leading to the acene units.7,12
Isomer 1′ (violet rectangles) results from the cyclisation of the minor isomer 2b (Fig. 1a and b). Tetracene dimers 6 and 7 (yellow and green rectangles) correspond to formal products of hydrogenolysis of the C-Br bonds of 2a/2b. In order to confirm the structure of all the resulting products we attached CO molecule to the microscope tip apex and applied the so-called bond-resolved STM (BR-STM).13 Comparison of the standard high resolution STM with BR-STM provides unequivocal identification of the structure of 1 (CBDT, Fig. 1c and d), as well as 1′, 6, 7 (Fig. S1, ESI†). DFT calculations were performed to analyse the interaction of the molecule 1 (CBDT) with the Au(111) surface (Fig. 1e). The molecule adopts an almost planar conformation with the mean surface to molecule distance of 3.31 Å, preferentially oriented along the [−110] (or equivalent) direction of the surface, in accordance with the experimental observation.
While 1 (CBDT) has been already obtained among other products by the surface-promoted formal [2+2] cycloaddition of the 2,3-dibromotetracene precursors on Ag(111),6b we describe its detailed electronic properties on surface for the first time. Thus, we have applied single point STS measurements to determine the energetic position of the resonances combined with the spatial mapping of the electronic clouds and corroborated by DFT calculations (Fig. 2). The single point dI/dV STS data acquired over 1 (CBDT) is shown in Fig. 2a. The lateral tip position during acquisition of the data is marked by coloured dots within the insets. The occupied state part of the spectrum, displayed in blue, contains two well-defined resonances centred at approximately −1.25 V and −0.88 V, labelled as HOMO−1 and HOMO, respectively. The empty state measurements provide two other resonances captured at approximately 1.25 V and 2.1 V assigned to the LUMO and LUMO+1, respectively. In order to reach deeper understanding of the electronic structure we performed spatial dI/dV mapping and corroborated the results with DFT calculations. Upper panels of Fig. 2b show the dI/dV STS images recorded at voltages corresponding to the resonances shown in Fig. 2a. In the bottom of Fig. 2b, the corresponding calculated dI/dV images are shown. From the comparison, which show reasonable agreement, it seems evident the resonances at −1.25 V and −0.88 V are dominated by HOMO−1 and HOMO, respectively, whereas the empty state recorded at +1.25 V could be linked with LUMO. The dI/dV map recorded at +2.1 V does not provide intense intramolecular features, however, as expected from the calculations, the signal reveals higher intensity at the molecule termini, therefore it is reasonable to assign the resonance as originating from LUMO+1. This is further supported by the analysis of the calculated projected density of states (PDOS) displayed for the molecule on Au(111) (Fig. 2c). The data clearly show the empty state maxima separated by approximately 0.7 eV, in accordance with the experiment. Similarly, the filled state part of the PDOS shows the presence of two peaks close to Fermi level and separated by approximately 0.3 eV, which is in agreement with experimentally observed interval. The calculations indicate a slight charge transfer between the molecule and the substrate resulting in a 0.2-0.3 eV shift of molecular states, whereas the hybridization of the molecular orbitals of 1 (CBDT) with the underlying Au(111) substrate is rather weak. The weak molecule–surface interaction is also supported by the energetics of the system. The calculated binding energy is 3.67 eV.
 | ||
| Fig. 2 Electronic structure of 1 (CBDT) on Au(111). (a) single point dI/dV STS data of 1 (CBDT), the coloured dots in the insets show the lateral position of the microscope tip during data acquisition; (b) experimental (top) and calculated (bottom) dI/dV images of 1 (CBDT); tunnelling current: 150 pA, scale bar: 0.3 nm (c) calculated density of states projected on 1 (CBDT, blue line, magnified 5 times) and on top-layer Au atoms (red line). | ||
The STS measurements indicate that the gap value for 1 (CBDT) on Au(111) reaches approximately 2.13 eV. The value is, as expected from the inclusion of the cyclobutadiene ring, much higher than the transport gap of 1.2 eV determined for nonacene.7 In fact, it is more similar to the values obtained for pentacene (2.2 eV)14 or tetracene (2.9 eV).15
The 36 π-electron system of 1 (CBDT) may be viewed as formally antiaromatic resulting from [4n]annulene resonance contributions (Scheme 3, I and II), notably those associated with the inner cyclobutadiene ring (ring A). However, cyclobutadiene-like conjugation is easily disrupted by ortho-fusion of aromatic subunits, as in biphenylene,16 and dibenzo[b,h]biphenylene.8d1 (CBDT) can thus also be described as consisting of two independent 18 π-electron tetracene subunits with limited conjugation across the four-membered ring (Scheme 3, III). A radialene-like contribution2 may also be envisaged (Scheme 3, IV), in which two anthracene subunits surround the central [4]radialene core.
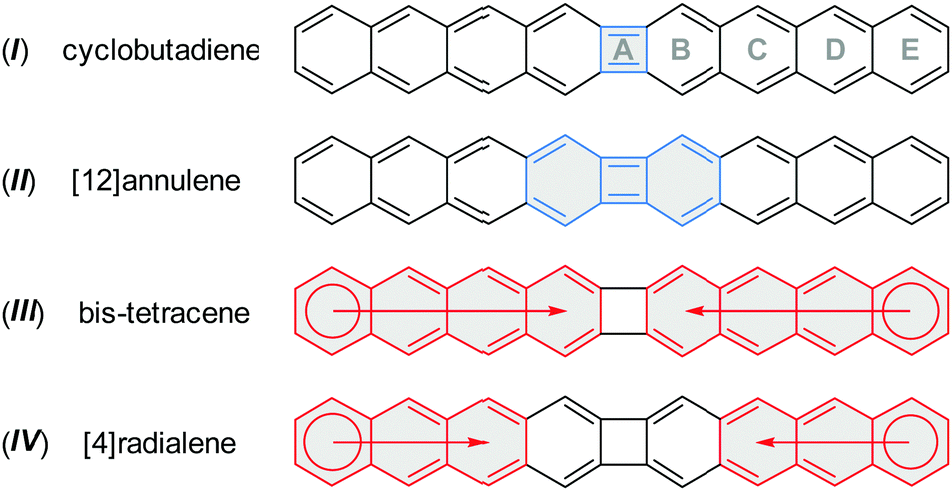 | ||
| Scheme 3 Selected possible contributions to π-conjugation in 1 (CBDT). | ||
Relative contributions of these different conjugation modes were probed using theoretical methods. The geometry of 1 (CBDT) obtained from density functional theory (DFT) calculations reveals a bond length pattern consistent with a dominant [4]radialene contribution (Fig. S3 and Table S1, ESI†). In particular, all C–C bonds in the four-membered ring are significantly elongated (1.460 and 1.484 Å), corresponding to Pauling bond orders17n = 1.26 and 1.18, respectively. In contrast, the formal double bond of the [4]radialene substructure has a length of 1.355 Å (n = 1.70), consistent with significant localization.
Nucleus-independent chemical shift18 (NICS) calculations shows a paratropic current in the four-membered ring of 1 (CBDT, Fig. 3a and b).19 The NICS(1) value calculated above the centre of the latter ring is relatively low (2.71 ppm), indicating that the local cyclobutadiene-like antiaromaticity is indeed diminished. The NICS(1) values in adjacent six-membered rings are higher than in the outer anthracene sections of the molecule (−6.02 for ring Bvs. ca. −10 to −13 ppm for rings C–E, respectively). These features agree with the behaviour predicted for lower linear diacenocyclobutanes.8d The aromatic character of rings B in all these systems was also found to be reduced relative to the outer sections of the acene substructures. The above findings are further confirmed by anisotropy of induced current density20 (ACID) calculations (Fig. 3c). A strong diatropic (clockwise) current is observed in each of the outer anthracene moieties (rings C–E), contrasting with the much weaker circulation observed in ring B. A weak paratropic current is identified in the four-membered ring A, again confirming partial cyclobutadiene character of 1 (CBDT).
 | ||
| Fig. 3 2D nucleus-independent chemical shift (NICS) scans performed (A) 1 Å above the molecular plane of 1 (CBDT) and (B) perpendicular to the molecular plane (GIAO/B3LYP/6-31G(d,p)). NICS(1) shifts evaluated above ring centers are indicated in panel B. (C) Anisotropy of induced current density (ACID) plot (CSGT/B3LYP/6-31G(d,p), π-only, 0.03 isovalue). Expansions show principal dia- and paratropic currents (red and blue arrows, respectively). | ||
In conclusion, by the combination of solution and on-surface chemistry we have generated the formally antiaromatic analogue of nonacene – the cyclobuta[1,2-b:3,4-b′]ditetracene 1 (CBDT) on Au(111). The structure of the surface-assisted reaction products has been in detail analysed by the BR-STM measurements with the microscope tip functionalized by a CO molecule. The electronic structure of the generated molecule has been visualized by STS measurements corroborated by theoretical modelling. The analysis indicates that the transport band gap of the 1 (CBDT) on Au(111) reaches approximately 2.13 eV, which is almost 1 eV larger than for the fully aromatic nonacene. Theoretical calculations indicate that π conjugation is dominated by a radialene contribution, with an admixture of cyclobutadiene-like antiaromaticity.
We acknowledge financial support from the National Science Center, Poland (2017/26/E/ST3/00855; 2018/29/B/ST5/01572), the SciMat Priority Research Area budget under the programme Excellence Initiative – Research University at the Jagiellonian University (U1U/P05/NW/03.10), ERC Proof of Concept GAP 837225, MCIN/AEI/10.13039/501100011033 (PID2019-104815GB-I00 and CEX2019-000925-S). Quantum chemistry calculations were performed at the Faculty of Mathematics, Physics and Computer Science of the Maria Curie-Sklodowska University and at the Wrocław Center for Supercomputing.
Conflicts of interest
There are no conflicts to declare.References
- J. E. Anthony, Angew. Chem., Int. Ed., 2008, 47, 452–483 CrossRef CAS PubMed.
- (a) R. R. Parkhurst and T. M. Swager, J. Am. Chem. Soc., 2012, 134, 15351–15356 CrossRef CAS PubMed; (b) P. Biegger, M. Schaffroth, O. Tsverkoy, F. Rominger and U. H. F. Bunz, Chem. – Eur. J., 2016, 22, 15896–15901 CrossRef CAS PubMed.
- (a) B. C. Berris, G. H. Hovakeemian, Y. H. Lai, H. Mestdagh and K. P. C. Vollhardt, J. Am. Chem. Soc., 1985, 107, 5670–5687 CrossRef CAS; (b) M. Hirthammer and K. P. C. Vollhardt, J. Am. Chem. Soc., 1986, 108, 2481–2482 CrossRef CAS PubMed; (c) R. H. Schmidt-Radde and K. P. C. Vollhardt, J. Am. Chem. Soc., 1992, 114, 9713–9715 CrossRef CAS; (d) C. Eickmeier, D. Holmes, H. Junga, A. J. Matzger, F. Scherhag, M. Shim and K. P. C. Vollhardt, Angew. Chem., Int. Ed., 1999, 38, 800–804 CrossRef CAS; (e) S. Han, A. D. Bond, R. L. Disch, D. Holmes, J. M. Schulman, S. J. Teat, K. P. C. Vollhardt and G. D. Whitener, Angew. Chem., Int. Ed., 2002, 41, 3223–3227 CrossRef CAS; (f) O. S. Miljanic, K. P. C. Vollhardt, M. M. Haley and R. R. Tykwinski, Carbon-Rich Compounds: From Molecules to Materials, Wiley-VCH, Weinheim, 2006 Search PubMed.
- (a) Z. Jin, Y. C. Teo, N. G. Zulaybar, M. D. Smith and Y. Xia, J. Am. Chem. Soc., 2017, 139, 1806–1809 CrossRef CAS PubMed; (b) Z. Jin, Z.-F. Yao, K. P. Barker, X. Pei and Y. Xia, Angew. Chem., Int. Ed., 2019, 58, 2034–2039 CrossRef CAS PubMed.
- J. Wang, M. Chu, J.-X. Fan, T. K. Lau, A.-M. Ren, X. Lu and Q. Miao, J. Am. Chem. Soc., 2019, 141, 3589–3596 CrossRef CAS PubMed.
- (a) M. Koch, M. Gille, S. Hecht and L. Grill, Surf. Sci., 2018, 678, 194–200 CrossRef CAS; (b) C. Sánchez-Sánchez, A. Nicolai, F. Rossel, J. Cai, J. Liu, X. Feng, K. Müllen, P. Ruffieux, R. Fasel and V. Meunier, J. Am. Chem. Soc., 2017, 139, 17617–17623 CrossRef PubMed; (c) B. V. Tran, T. A. Pham, M. Grunst, M. Kivala and M. Stöhr, Nanoscale, 2017, 9, 18305–18310 RSC; (d) C. Sánchez-Sánchez, T. Dienel, A. Nicolai, N. Kharche, L. Liang, C. Daniels, V. Meunier, J. Liu, X. Feng, K. Müllen, J. R. Sánchez-Valencia, O. Gröning, P. Ruffieux and R. Fasel, Chem. – Eur. J., 2019, 25, 12074–12082 CrossRef PubMed.
- R. Zuzak, R. Dorel, M. Krawiec, B. Such, M. Kolmer, M. Szymonski, A. M. Echavarren and S. Godlewski, ACS Nano, 2017, 11, 9321–9329 CrossRef CAS PubMed.
- (a) S. Clair and D. G. de Oteyza, Chem. Rev., 2019, 119(7), 4717–4776 CrossRef CAS PubMed; (b) L. Grill, M. Dyer, L. Lafferentz, M. Persson, M. V. Peters and S. Hecht, S. Nat. Nanotechnol., 2007, 2, 687–691 CrossRef CAS PubMed; (c) J. Cai, P. Ruffieux, R. Jaafar, M. Bieri, T. Braun, S. Blankenburg, M. Muoth, A. P. Seitsonen, M. Saleh, X. Feng, K. Müllen and R. Fasel, Nature, 2010, 466, 470–473 CrossRef CAS PubMed; (d) S. Kawai, K. Takahashi, S. Ito, R. Pawlak, T. Meier, P. Spijker, F. P. Canova, J. Tracey, K. Nozaki, A. S. Foster and E. Meyer, ACS Nano, 2017, 11, 8122–8130 CrossRef CAS PubMed.
- C. Gu, D. Zhu, M. Qiu, L. Han, S. Wen, Y. Lia and R. Yang, New J. Chem., 2016, 40, 7787–7794 RSC.
- R. Dorel, P. R. McGonigal and A. M. Echavarren, Angew. Chem., Int. Ed., 2016, 55, 11120–11123 CrossRef CAS PubMed.
- (a) R. Zuzak, J. Castro-Esteban, P. Brandimarte, M. Engelund, A. Cobasm, P. Piatkowski, M. Kolmer, D. Pérez, E. Guitián, M. Szymonski, D. Sánchez-Portal, S. Godlewski and D. Peña, Chem. Commun., 2018, 54, 10256–10259 RSC; (b) S. Mishra, M. Krzeszewski, C. A. Pignedoli, P. Ruffieux, R. Fasel and D. T. Gryko, Nat. Commun., 2018, 9, 1714 CrossRef PubMed.
- R. Dorel and A. M. Echavarren, Eur. J. Org. Chem., 2017, 14–24 CrossRef CAS PubMed.
- (a) S. Song, J. Su, X. Peng, X. Wu and M. Telychko, Surf. Rev. Lett., 2021, 28, 214007 Search PubMed; (b) G. D. Nguyen, H.-Z. Tsai, A. A. Omrani, T. Marangoni, M. Wu, D. J. Rizzo, G. F. Rodgers, R. R. Cloke, R. A. Durr, Y. Sakai, F. Liou, A. S. Aikawa, J. R. Chelikowsky, S. G. Louie, F. R. Fischer and M. F. Crommie, Nat. Nanotechnol., 2017, 12, 1077–1082 CrossRef CAS PubMed; (c) J. I. Urgel, S. Mishra, H. Hayashi, J. Wilhelm, C. A. Pignedoli, M. Di Giovannantonio, R. Widmer, M. Yamashita, N. Hieda, P. Ruffieux, H. Yamada and R. Fasel, Nat. Commun., 2019, 10, 861 CrossRef PubMed.
- W.-H. Soe, C. Manzano, A. De Sarkar, N. Chandrasekhar and C. Joahim, Phys. Rev. Lett., 2009, 102, 176102 CrossRef PubMed.
- F. Eisenhut, T. Kühne, F. García, S. Fernández, E. Guitián, D. Pérez, G. Triquier, G. Cuniberti, C. Joahim, D. Peña and F. Moresco, ACS Nano, 2020, 14, 1011–1017 CrossRef CAS PubMed.
- W. C. Lothrop, J. Am. Chem. Soc., 1941, 63, 1187–1191 CrossRef CAS.
- L. Pauling, J. Am. Chem. Soc., 1947, 69, 542–553 CrossRef CAS.
- Z. Chen, C. S. Wannere, C. Corminboeuf, R. Puchta and P. von R. Schleyer, Chem. Rev., 2005, 105, 3842–3888 CrossRef CAS PubMed.
- (a) K. Frederickson, L. N. Zakharov and M. M. Haley, J. Am. Chem. Soc., 2016, 138, 16827–16838 CrossRef PubMed; (b) R. Ayub, O. El Bakouri, K. Jorner, M. Solà and H. Ottosson, J. Org. Chem., 2017, 82, 6327–6340 CrossRef CAS PubMed.
- D. Geuenich, K. Hess, F. Köhler and R. Herges, Chem. Rev., 2005, 105, 3758–3772 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d2cc00479h |
| This journal is © The Royal Society of Chemistry 2022 |