
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Titanium isopropoxide-mediated cis-selective synthesis of 3,4-substituted butyrolactones from CO2†
Aleksi
Sahari
 a,
Cuong Dat
Do
b,
Jere K.
Mannisto
a,
Emanuele
Antico
a,
Angelo
Amaratunga
a,
Kathrin H.
Hopmann
*c and
Timo
Repo
*a
a,
Cuong Dat
Do
b,
Jere K.
Mannisto
a,
Emanuele
Antico
a,
Angelo
Amaratunga
a,
Kathrin H.
Hopmann
*c and
Timo
Repo
*a
aDepartment of Chemistry, University of Helsinki, FI-00014 University of Helsinki, Finland. E-mail: timo.repo@helsinki.fi
bHylleraas Center for Quantum Molecular Sciences, Department of Chemistry, UiT The Arctic University of Norway, 9037 Tromsø, Norway
cDepartment of Chemistry, UiT The Arctic University of Norway, 9037 Tromsø, Norway. E-mail: kathrin.hopmann@uit.no
First published on 14th February 2022
Abstract
We report a Ti(OiPr)4-mediated multicomponent reaction, which produces 3,4-substituted cis-δ-lactones from alkyl magnesium chloride, benzaldehyde and CO2. The key intermediate, titanacyclopropane, is formed in situ from Ti(OiPr)4 and a Grignard reagent, which enables 1,2-dinucleophilic reactivity that is used to insert carbon dioxide and an aldehyde. An alternative reaction route is also described where a primary alkene is used to create the titanacyclopropane. A computational analysis of the elementary steps shows that the carbon dioxide and the aldehyde insertion proceeds through an inner-sphere mechanism. A variety of cis-butyrolactones can be synthesized with up to 7![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 diastereoselectivity and 77% yield.
:1 diastereoselectivity and 77% yield.
Carbon dioxide is a greenhouse gas and a renewable, non-toxic and inexpensive C1-source; therefore, its utilization in chemical synthesis is desirable. Typically, organometallic species are necessary to generate C–CO2 bonds.1 Grignard reagents and organolithium reagents are well known to react as nucleophiles towards CO2 to form the corresponding metal carboxylates.2 In this context, we envisioned organotitanium chemistry to provide an opportunity for alternative reactivity with CO2. Titanium(IV)alkoxides mediate reductive elimination of Grignard and alkyl lithium reagents to form highly reactive titanacyclopropanes (Scheme 1A). These species have been employed in the synthesis of cyclopropanols and cyclopropyl amines in the Kulinkovich reaction (Scheme 1B).3 There, the 1,2-dinucleophilic titanacyclopropane reacts with the formally dielectrophilic ester or amide in two consequent steps. Electrophiles other than esters and amides are also reactive towards the titanacyclopropane ring: CO2, I2, H2O and a variety of organic electrophiles can readily insert into the Ti–C bond.3a,4
 | ||
| Scheme 1 Key examples of titanacyclopropane reactivity.3a,4c | ||
However, in most of these applications, the dinucleophilic nature of the titanacyclopropane is not completely utilized and the second Ti–C bond is usually quenched with acid,5 rather than using it to insert another functionality.
Rassadin and Six demonstrated that the insertion of CO2 into titanacyclopropane results in a five-membered titanalactone (Scheme 1C), which does not react with another CO2 molecule.4c The remaining Ti–C bond can be cleaved with water or by I2, NBS or O2 to yield β-substituted carboxylic acids.
We predicted that a carbon electrophile may be able to insert into the titanalactone ring (Scheme 1D). This would generate two carbon–carbon bonds from cheap and simple starting materials, providing a useful method to rapidly increase complexity starting from simple materials. Similar insertions have been observed with titanium(II)–alkyne complexes,6 but there are limited examples for corresponding titanium(II)–alkene complexes. Previously Eisch et al. had reported a three- component reaction of ethene, phenyl nitrile and CO2 with preformed Ti(OiPr)2,7 but its reactivity was not further explored.
Initially, we used butyl lithium to form the titanacyclopropane, which was reacted sequentially with CO2 and p-fluorobenzaldehyde. After optimization (Table 1), up to 77% yield of cis-lactone could be obtained, in addition to 11% of trans-lactone. The generated titanacyclopropane seems to decompose quickly under these reaction conditions. Therefore it was critical to have precise control of temperature and duration of each step. Under non-optimized conditions, titanium-derived decomposition products can further react with the aldehyde to produce a myriad of unwanted side products via for example pinacol coupling or McMurry reaction.
|
|
||
|---|---|---|
| # | Deviation from | Yieldb (%) |
|
a Optimized conditions: 2.6 mmol BuLi, 1.3 mmol Ti(OiPr)4, 1 mmol p-fluorobenzaldehyde, 3 ml of THF. Step 1: 1 min at 0 °C, step 2: 5 min CO2 flush at 0 °C followed by 25 min at 20 °C. Step 3 reaction time 3 days at 20 °C.
b Entries 1 and 2 are NMR yield and entries 3–10 are calibrated GC yield.
c Irreproducible results.
d Ratio of BuLi:Ti(OiPr)4:p-fluorobenzaldehyde.
|
||
| 1 | None | 77 |
| 2 | Step 1: 2 min | 63 |
| 3 | Step 1: 0.5 min | 14–56c |
| 4 | Step 1: 5 min | 53 |
| 5 | Step 1: 5 min, step 2: 15 min | 44 |
| 6 | Step 1: 5 min, step 2: 2 h | 39 |
| 7 | Step 1: 5 min, step 2: 2 h, Et2O as solvent | 19 |
| 8 | Step 1: 5 min, step 2: 2 h, stoichiometry 2.1:1.05:1d |
26 |
| 9 | As entry 8, but 20 °C throughout all steps | Trace |
| 10 | Step 3: 40 °C, 16 h | 68 |
| 11 | With LiCl (2.6 eq.) | 71 |

After proving the feasibility of the reaction using butyl lithium, we proceeded to study the reactivity with Grignard reagents, since they have better availability and are more convenient to make. When butyl magnesium chloride was used to make the titanacyclopropane, its decomposition seemed to be faster compared to the one generated from butyl lithium. The temperature for step 1 was therefore reduced from 0 °C to −20 °C and 2.6 eq. of LiCl was added. With these modifications, 73% yield of cis-lactone could be obtained. The effect of lithium chloride was investigated by adding a solution of LiCl in THF to the mixture right before each step (see Table S5, ESI†). LiCl greatly improves the reaction yield when added before step 1 (from 31% to 65% yield) but has almost no effect when added after step 1. LiCl is known to break up polymeric aggregates of Grignard reagents by forming RMgCl·LiCl complexes, which can increase the kinetic reactivity.8 LiCl had no significant effect on the reaction involving BuLi (Table 1, entry 11), which would further suggest that its only function is to increase the reactivity of the Grignard reagent.
We also evaluated the scope of Grignard reagents (Scheme 2). Simple Grignard reagents worked well, but many functionalities were unstable under the reaction conditions. For example, ether substituents undergo elimination and do not yield any product (see Fig. S1, ESI†). Secondary Grignard reagents, such as isopropyl and c-HexMgCl, only gave direct addition products to CO2 or aldehyde. Since the formation of the titanacyclopropane is a critical step in the reaction, further improvements could likely be obtained by individually optimizing the conditions for each Grignard reagent.
 | ||
| Scheme 2 Lactone formation from a variety of Grignard reagents and aldehydes. Yield is single cis-diastereomer yield measured by 1H-NMR with 1,3,5-trimethoxybenzene as internal standard. Isolated yield for the cis-diastereomer is in brackets. Stoichiometry is 2.6 mmol BuMgCl, 2.6 mmol LiCl, 1.3 mmol Ti(OiPr)4, 1 mmol aldehyde, 3 ml of THF. a69% 18O-content starting from 76% 18O-p-fluorobenzaldehyde. btrans-Diastereomer yield, which is the major product. | ||
Next, we studied different electrophiles in step 3 in a reaction based on BuMgCl. Various benzaldehydes and α,β-unsaturated cinnamaldehyde provided the desired lactone (Scheme 2). Among the tested substrates, epoxides, imines, nitriles, ketones and aliphatic aldehydes were non-reactive (Fig. S2 and S3, ESI†). The more reactive acrolein and iminiums only formed various side products, which were not further analyzed.
With benzaldehydes (Scheme 2), the reaction tolerates a variety of common functionalities with both electron donating (11jp-OMe 58%) and electron withdrawing (11mp-COOMe 58%, 11q Br 55%) properties. Note that ester 11m and nitrile 11n can potentially react with organometallic reagents, but are tolerated here. However, the reaction has a strong dependence on the substitution pattern, where m-MeO (11k 24%) and o-MeO (11l 18%) yield less product than p-MeO (11j). The aldehydes are completely consumed in the reaction and the variation in yield is explained by selectivity between aldehyde insertion to the titanalactone or side reactions, such as pinacol coupling. The cis-diastereomer is the favoured product with a 7–5:1 ratio, although accurate determination could only be done for fluorinated compounds with 19F-NMR, due to overlapping impurities in 1H for the trans-diastereomer. An exception to this selectivity was 11o, where the trans-diastereomer was the major product. However, when BuLi was used to make 11o 20% trans and 21% cis crude was obtained. The crude quickly isomerises to >10:1 trans-diastereomer on silica (see ESI† for details). This isomerization behaviour was not detected for any other lactones. The 18O labelled 11g shows that the oxygen in the structure originates from the benzaldehyde.
In the Kulinkovich type reactions, it has been shown that if a primary alkene or alkyne is added to the mixture, the reaction will exchange the Grignard reagent with the added unsaturated compound.4a,9 Therefore, we investigated primary alkenes in our reaction.10 When c-HexMgCl was used as the Grignard reagent, we could incorporate 1-hexene to yield 34% of cis-lactone after optimization (Scheme 3). Cyclic Grignard reagents are preferred, since they form a less stable titanacyclopropane, which facilitates the exchange with the primary alkene (Tables S7–8 and Fig. S11, ESI†). Primary Grignard reagents and Buli produce a mixture of the exchanged and non-exchanged product (Table S8, ESI†). Around 30% yield could be obtained for several alkenes (Scheme 3), however vinyl butyl ether and styrene did not produce any lactone. This is likely due to the polarization of the double bond. Gratifyingly, the reaction tolerated trimethyl silyl and alkyl bromide groups, which were difficult substrates in the primary alkyl Grignard reagent version of this reaction (Scheme 2).
 | ||
| Scheme 3 Lactone formation from alkenes. Yield is single cis-diastereomer yield measured by 1H-NMR with 1,3,5-trimethoxybenzene as internal standard, isolated yield is in brackets. Stoichiometry is 4 mol alkene, 2.6 mmol c-HexMgCl, 1.3 mmol Ti(OiPr)4, 1 mmol p-fluorobenzaldehyde, 3 ml of THF. | ||
The mechanistic details of the transformation of dialkyltitanium to titanacyclopropane 3 have recently been investigated computationally by Bertus (Fig. S8, ESI†).11 We have here employed DFT methods to investigate the reaction of titanacyclopropane 3a (Fig. S9, ESI†) with CO2, and the subsequent benzaldehyde insertion leading to the formation of 3,4-substituted butyrolactones (see ESI† for detailed protocol).
At the preferred inner sphere transition state for CO2 insertion into 3a (TSAB, Fig. 1), the terminal carbon of butene performs a nucleophilic attack on the CO2 carbon, with a barrier of only 2.2 kcal mol−1 relative to A (Fig. 2, 298 K). Attack of the internal butene carbon has a slightly higher barrier (2.8 kcal mol−1). The barrier for an alternative outer sphere CO2 insertion is 24.8 kcal mol−1 (298 K, Fig. S10, ESI†), demonstrating that the inner sphere CO2 insertion is strongly preferred. Similar results have been obtained for the Kulinkovich reaction, where the first insertion of ester proceeds through a low-barrier inner sphere pathway.12 The carboxylation barrier computed here is lower than what may be expected from the reaction time (30 minutes warmed from −20 °C to 20 °C), however, we speculate that the increased reaction time is needed to decompose reactive organometallic species that would compete to consume the aldehyde in the next step.
 | ||
| Fig. 1 Preferred inner-sphere transition state geometry for insertion of CO2 into 3 (TSAB, B3LYP-D3/6-31+G(d,p), CPCM[THF]). | ||
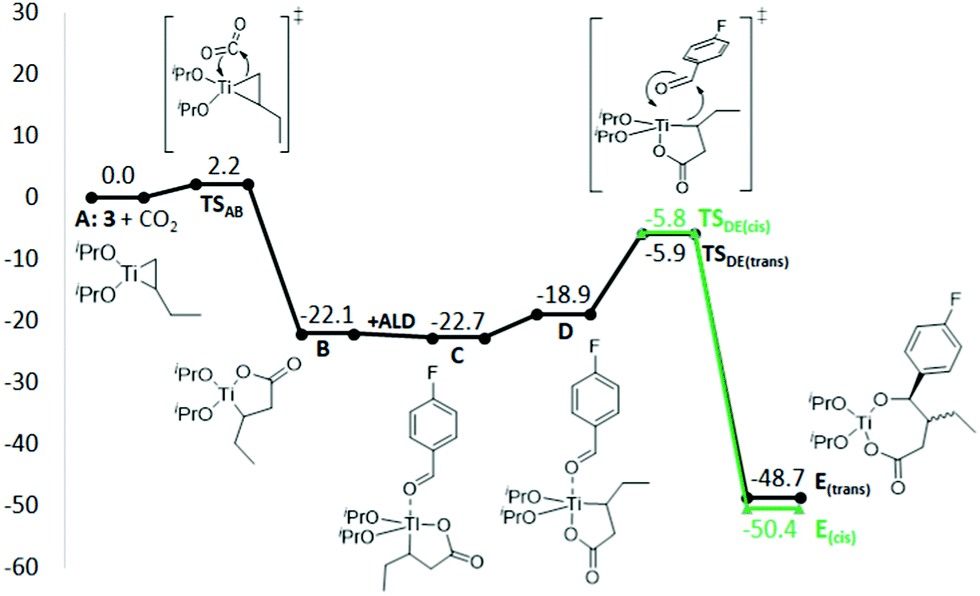 | ||
| Fig. 2 Gibbs free energy profile for conversion of titanacyclopropane 3 to titanalactone B, followed by aldehyde (ALD) insertion to form E (kcal mol−1, 298 K, B3LYP-D3/def2-TZVP,CPCM[THF]//B3LYP-D3/6-31+G(d,p),CPCM[THF]). | ||
The transformation of titanalactone B to butyrolactones starts with the coordination of 4-fluorobenzaldehyde to form C, which has a distorted trigonal bipyramidal geometry, with the aldehyde trans to the Ti–C bond. The alternative cis geometry D is 3.8 kcal mol−1 less stable (298 K), but it needs to be formed to allow for subsequent aldehyde insertion viaTSDE (Fig. 2). TSDE has two possible conformations, leading to either E(trans) or E(cis), with the latter being thermodynamically preferred. The experimentally observed 7:1 cis selectivity corresponds to ca. 1 kcal mol−1 difference in energy barrier, which is within the error of the computational approach.
The computed barriers for insertion of acetophenone and phenylmethanimine into B are 20.9 and 28.6 kcal mol−1, respectively (Table S9, ESI†), compared to 16.8 kcal mol−1 for 4-fluorobenzaldehyde (298 K). The higher barriers are due to a stronger binding of ketone and imine to titanium compared to the aldehyde and are in line with the experimentally observed lack of reactivity for the former two substrates, although the overall barrier for acetophenone indicates that some reactivity may have been expected.
Finally, we also studied the mechanistic details of the alkene exchange reaction (Scheme 3). Given that the direct decoordination of an alkene from species A is thermodynamically highly disfavoured (28.4 or 35.0 kcal mol−1 depending on the alkene, see Fig. S11 and S12, ESI†), the reaction most likely proceeds through an associative mechanism involving formation of an intermediate, where both the alkene originating from the Grignard reagent (cyclohexene) and the added alkene (1-hexene) coordinate simultaneously (Fig. S11, ESI,†S15CyHex). After the alkene originating from the Grignard reagent decoordinates, a new species A is obtained, and the reaction follows the standard mechanism (Fig. 2). Calculations with cyclohexene as the original alkene and 1-hexene as the added alkene show a 9.9 kcal mol−1 barrier to decoordinate 1-hexene from species S15CyHex, compared to 2.3 kcal mol−1 for cyclohexene, which is in line with experimental results showing that an alkene exchange takes place under these conditions (Scheme 3, 11d). The exchange is driven by the greater stability for coordinating 1-hexene than cyclohexene (−6.3 kcal mol−1, Fig. S11, ESI†). The analogous coordination of CO2 to species A was also calculated, and the obtained barrier (16.7 kcal mol−1) is much higher than that of the inner sphere insertion (2.2 kcal mol−1). This shows that CO2 will rather insert itself directly than first coordinate to species A (Fig. S13, ESI†).
In summary, we have demonstrated a CO2-based method for the cis-selective synthesis of 3,4 butyrolactones using simple and easily available materials. The reaction forms two new carbon–carbon bonds in a three-component reaction, which makes it a powerful synthetic tool. While unstable titanium- intermediates limit the scope of substrates, this method can be useful to create a substitution pattern that usually requires several additional synthetic steps. The computational analysis indicates low stability of titanacyclopropane and a high stability of titanalactone. Coordination of an electrophile (e.g. aldehyde, imine, or ketone) leads to a low-energy intermediate, which determines the barrier of the final step. We foresee that the scope of the reaction might be expanded by changes to the steric and electronic properties of the initial titanium complex.
This work has been supported by CHEMS – The Doctoral Programme in Chemistry and Molecular Sciences at University of Helsinki, and the members of the Nordic Consortium for CO2 Conversion (NordCO2). KHH and DDC thank the Research Council of Norway (Grant No. 300769 and 262695), the Tromsø Research Foundation (Grant No. TFS2016KHH), NordForsk (Grant No. 85378) and Notur – The Norwegian Metacenter for Computational Science (No. nn9330k and nn4654k).
Conflicts of interest
There are no conflicts to declare.References
- (a) C. S. Yeung and V. M. Dong, J. Am. Chem. Soc., 2008, 130, 7826–7827 CrossRef CAS PubMed; (b) T. Leon, A. Correa and R. Martin, J. Am. Chem. Soc., 2013, 135, 1221–1224 CrossRef CAS PubMed; (c) Q. Liu, L. Wu, R. Jackstell and M. Beller, Nat. Commun., 2015, 6, 5933 CrossRef PubMed; (d) S.-S. Yan, Q. Fu, L.-L. Liao, G.-Q. Sun, J.-H. Ye, L. Gong, Y.-Z. Bo-Xue and D.-G. Yu, Coord. Chem. Rev., 2018, 374, 439–463 CrossRef CAS; (e) S. Wang and C. Xi, Chem. Soc. Rev., 2019, 48, 382–404 RSC.
- H. Gilman and R. H. Kirby, Org. Synth., 1925, 5, 75 CrossRef.
- (a) O. G. Kulinkovich, A. I. Savchenko, S. V. Sviridov and D. A. Vasilevski, Mendeleev Commun., 1993, 3, 230–231 CrossRef; (b) A. de Meijere, C. M. Williams, A. Kourdioukov, S. V. Sviridov, V. Chaplinski, M. Kordes, A. I. Savchenko, C. Stratmann and M. Noltemeyer, Chem. – Eur. J., 2002, 8, 3789–3801 CrossRef CAS PubMed; (c) O. G. Kulinkovich, S. V. Sviridov, D. A. Vasilevskii and T. S. Pritytskaya, Zh. Org. Khim., 1989, 25, 2244–2245 CAS.
- (a) F. Sato, H. Urabe and S. Okamoto, Chem. Rev., 2000, 100, 2835–2886 CrossRef CAS PubMed; (b) A. Wolan and Y. Six, Tetrahedron, 2010, 66, 15–61 CrossRef CAS; (c) V. A. Rassadin and Y. Six, Tetrahedron, 2014, 70, 787–794 CrossRef CAS.
- (a) V. K. Chenniappan and R. J. Rahaim, Org. Lett., 2016, 18, 5090–5093 CrossRef CAS PubMed; (b) P. Shao, S. Wang, C. Chen and C. Xi, Org. Lett., 2016, 18, 2050–2053 CrossRef CAS PubMed; (c) F. Cadoret, P. Retailleau and Y. Six, Tetrahedron Lett., 2006, 47, 7749–7753 CrossRef CAS; (d) O. L. Epstein, J. M. Seo, N. Masalov and J. K. Cha, Org. Lett., 2005, 7, 2105–2108 CrossRef CAS PubMed; (e) F. Sato and S. Okamoto, Adv. Synth. Catal., 2001, 343, 759–784 CrossRef CAS.
- (a) M. McLaughlin, M. Takahashi and G. C. Micalizio, Angew. Chem., Int. Ed., 2007, 46, 3912–3914 CrossRef CAS PubMed; (b) Y. Gao, M. Shirai and F. Sato, Tetrahedron Lett., 1997, 38, 6849–6852 CrossRef CAS.
- J. J. Eisch, J. N. Gitua, P. O. Otieno and X. Shi, J. Organomet. Chem., 2001, 624, 229–238 CrossRef CAS.
- (a) A. Krasovskiy and P. Knochel, Angew. Chem., Int. Ed., 2004, 43, 3333–3336 CrossRef CAS PubMed; (b) R. L. Bao, R. Zhao and L. Shi, Chem. Commun., 2015, 51, 6884–6900 RSC.
- (a) T. Nakagawa, A. Kasatkin and F. Sato, Tetrahedron Lett., 1995, 36, 3207–3210 CrossRef CAS; (b) Y. Six, Eur. J. Org. Chem., 2003, 1157–1171 CrossRef CAS.
- John J. Eisch, Adetenu A. Adeosun and John N. Gitua, Eur. J. Org. Chem., 2003, 4721–4727 CrossRef CAS.
- P. Bertus, Organometallics, 2019, 38, 4171–4182 CrossRef CAS.
- Y. D. Wu and Z. X. Yu, J. Am. Chem. Soc., 2001, 123, 5777–5786 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d2cc00446a |
| This journal is © The Royal Society of Chemistry 2022 |