
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The long-awaited synthesis and self-assembly of a small rigid C3-symmetric trilactam†
Yutang
Li
 a,
Daniel
Strand
a,
Stefan
Grimme
b,
Stefán
Jónsson
a and
Kenneth
Wärnmark
*a
a,
Daniel
Strand
a,
Stefan
Grimme
b,
Stefán
Jónsson
a and
Kenneth
Wärnmark
*a
aCentre for Analysis and Synthesis, Department of Chemistry, Lund University, SE-221 00 Lund, Sweden. E-mail: kenneth.warnmark@chem.lu.se
bMulliken Center for Theoretical Chemistry, Institut für Physikalische und Theoretische Chemie, Rheinische Friedrich-Wilhelms-Universität Bonn, 53115 Bonn, Germany
First published on 15th February 2022
Abstract
The challenging synthesis of a fused C3-symmetric trilactam (1) was executed in racemic and enantiomerically pure form. The rigidity, symmetry and high density of hydrogen bonding motifs make 1 an attractive candidate for self-assembly study, which revealed different hydrogen bond patterns in the crystals of rac-1-d3 and (+)-(SSS)-1.
As a non-covalent interaction, hydrogen bonding has received a lot of attention from chemists ever since its establishment in the early 1900s.1–3 Its directionality, predictability and tunability have made the hydrogen bond a powerful and reliable tool to manipulate structures and properties on a supramolecular level. Numerous molecules containing hydrogen bond donors and/or acceptors have been designed and synthesized as building blocks for host–guest chemistry4 or as supramolecular synthons for crystal engineering.5–8 One simple motif that is commonly found in these molecules is the amide group.9–11 Herein we report the first synthesis, characterization, and solid-state self-assembly of a remarkably small molecule containing three amide groups, trilactam 1 (Fig. 1), which was first proposed and modelled as a scaffold for supramolecular structures in 199312 and has been the target of synthetic efforts from several research groups at the latest since 1971.13
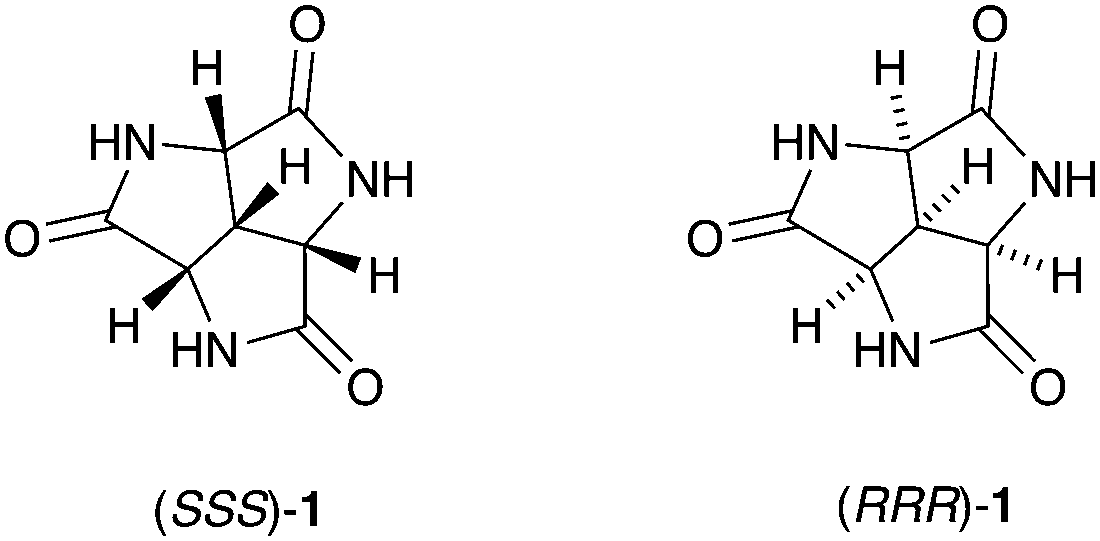 | ||
| Fig. 1 The structure of the racemic trilactam rac-1. | ||
The charm of 1 itself is revealed in many aspects, and one of them is its symmetry. From a purely aesthetic point of view, the structure of 1 possesses an attractive three-fold rotational symmetry which can be found both in Nature and human civilization.14,15 Furthermore, compound 1 is the smallest possible C3 symmetric fused trilactam to our best knowledge. Another attractive aspect is its chirality, which makes 1 a potential candidate for chiral recognition and asymmetric catalysis.14–16 In addition, the high density of amide groups in 1 also contribute to its versatility: The inherent cis amide groups provide three pairs of self-complementary hydrogen-bonding motifs within one molecule of 1. The rigid and planar nature of amides reinforces the molecular rigidity established by the concave tricyclic backbone. All these features make 1 an attractive candidate for hydrogen-bond based self-assembly. Some of the possible assemblies of 1 are modelled, which includes three regular closed-shell polyhedrons and a non-planar hexagonal tiling (Fig. S5, ESI†).17 The approximate cavity volumes of the three regular polyhedrons vary from 69 Å3 to 2298 Å3,18 which implies its versatility in host–guest chemistry. The hexagonal tiling illustrates the potential application of 1 in surface engineering.19–21 Moreover, there are several possible intermolecular binding modes (Fig. 2),17 which further increases the number of possible assemblies.
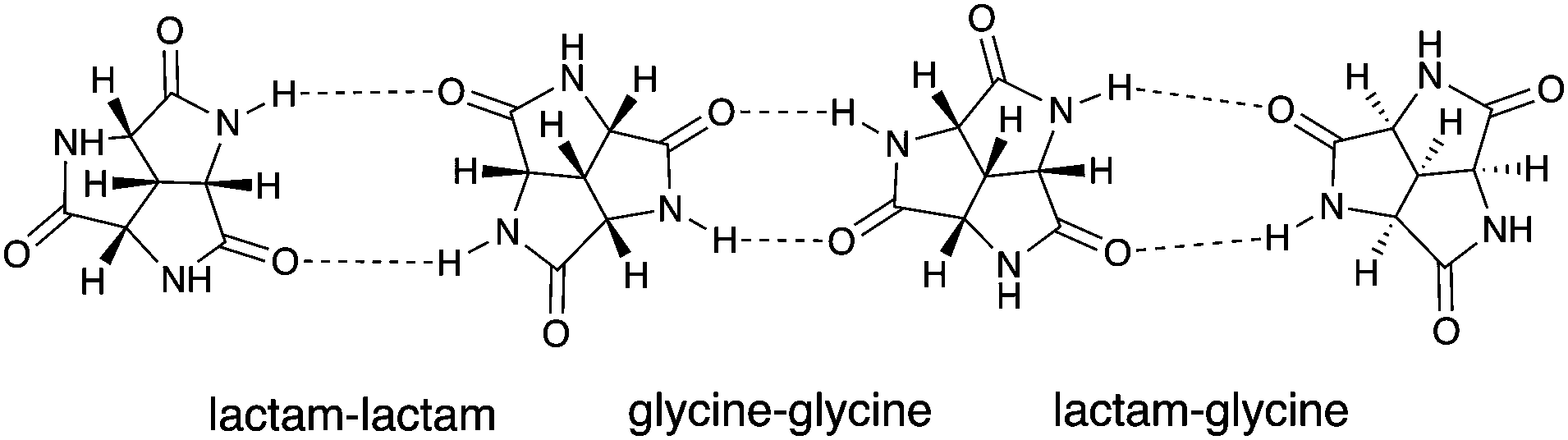 | ||
| Fig. 2 The three modes for (SSS)-1 to self-assemble via hydrogen bonding: lactam–lactam (left), glycine–glycine (middle) and lactam–glycine (right). | ||
Historically, synthetic efforts towards rac-1 have been carried out by several research groups following different strategies. One obvious and efficient synthetic strategy is to base the synthesis of rac-1 on its high symmetry. In 1971, Mock reported the synthesis of C3 symmetric triacetate rac-2 (Fig. 3a).13 It was later confirmed by Mock that his group initially prepared rac-2 as an intermediate in a planned synthesis of rac-1, where tri-substitution of rac-2 with azide nucleophile followed by azide reduction and concomitant ring closure of the corresponding tri(α-aminoacetate) would form rac-1.22 However, the substitution of rac-2 with sodium azide was found to trigger the elimination of HBr. Later the triacetate rac-3 (Fig. 3b) bearing no hydrogen at the central carbon was synthesized by Branda23 and Rebek with the expectation that this molecule would undergo the azide substitution.17 Unfortunately, the methyl group in rac-3 on the central carbon, although excluding the elimination of HBr, introduced neopentyl systems within the molecule, which hindered the reactivity of the bromides towards SN2 reactions with azide as a nucleophile.
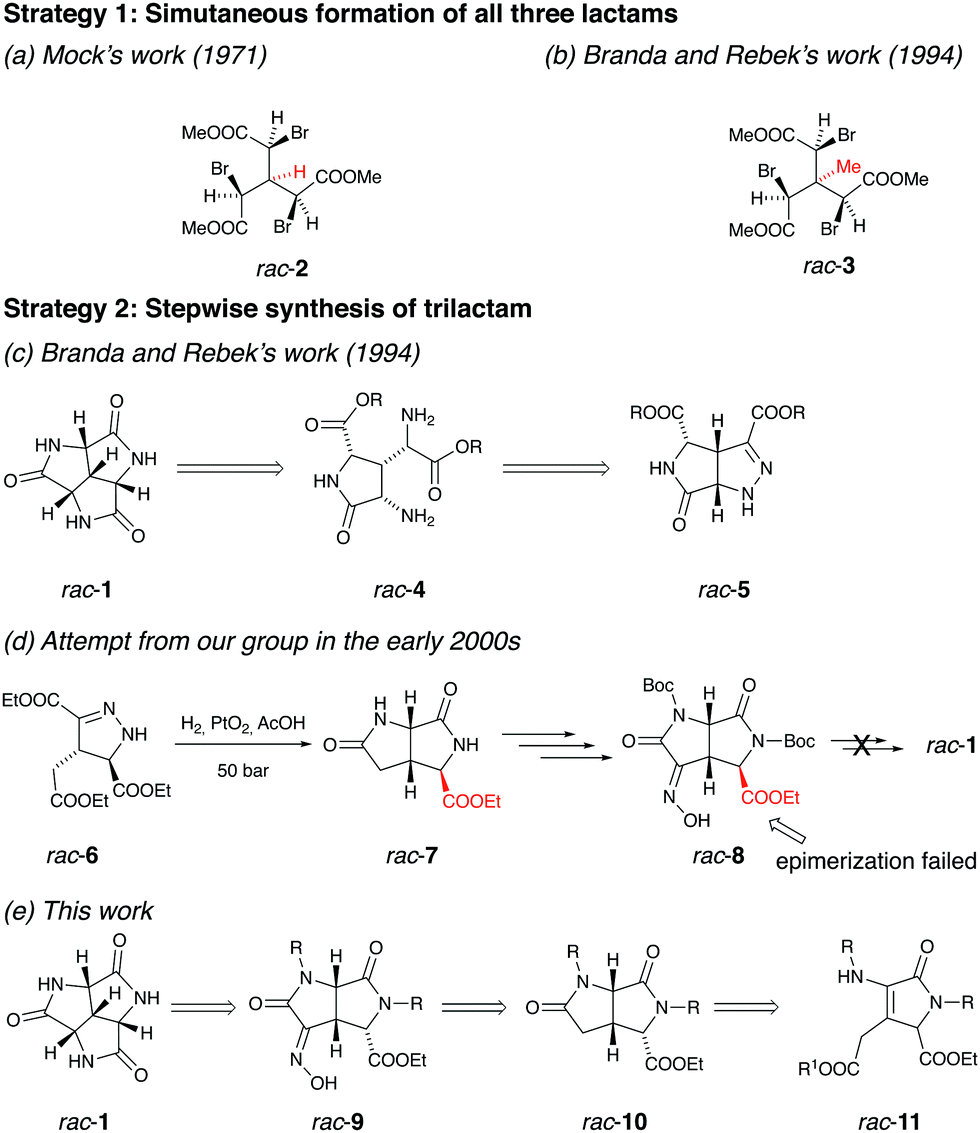 | ||
| Fig. 3 Synthetic efforts towards rac-1 using different strategies. | ||
Since the strategy based on the simultaneous formation of all three lactams failed, Branda and Rebek turned their attention to a stepwise procedure (Fig. 3c).17 The key intermediate in this route is bicyclic pyrazoline rac-5, which could afford rac-4via hydrogenation. Several synthetic routes were attempted in an endeavour to obtain rac-5 but turned out to be fruitless.
Unaware of the extensive efforts by Branda and Rebek, we started our journey towards rac-1 in the early 2000s aiming to study its self-assembly. Several different synthetic routes were attempted without succeeding to reach the target molecule. The furthest-reaching route is shown in Fig. 3d. Similar as planned in Branda and Rebek's second route, the reduction of pyrazoline rac-6 followed by the acid-promoted ring-closure gave dilactam rac-7, which was transformed to oxime rac-8 in several steps. Unfortunately, the obtained stereochemistry of the carboxylic ester in rac-8 prevented the final ring-closure. Epimerization of rac-8 to acquire the correct stereochemistry was attempted with no success.
A turning point appeared in 2018, when we were inspired by a three-component synthesis of highly functionalized 3-pyrrolin-2-ones reported by Palacios in the same year.24 We recognized that rac-11 could be a suitable synthon for rac-1. Based on this, we performed a new retrosynthetic analysis (Fig. 3e). The stereoselective hydrogenation of rac-11 followed by the ring-closure would afford dilactam rac-10, which possesses the correct stereochemistry to allow for the final ring-closure. The installation of the third nitrogen on rac-10 could be performed similarly as from rac-7 to rac-8 (Fig. 3d). The third lactam would be formed by the reduction of oxime rac-9 in acidic condition, followed by deprotection, giving the final trilactam rac-1.
As presented in Scheme 1, the synthesis of rac-1 started by the three-component synthesis of 3-pyrrolin-2-one rac-12, where p-methoxyphenyl (PMP) groups were installed on the amine and the lactam, respectively. Compared to the phosphoric acids that were used by Palacios in a similar cyclization,24 acetic acid was found to give a higher yield by facilitating the intramolecular lactamization. Treatment of rac-12 with pressurized hydrogen over palladium on activated charcoal allowed the stereoselective addition of hydrogen from the less sterically hindered face of the double bond. Further lactamization promoted by acetic acid established the cis-fused dilactam rac-13 with the desired stereochemistry of the carboxylic ester (Fig. S2, ESI†). The stepwise reaction was necessary, since direct hydrogenation in acetic acid resulted in a lower yield as well as the epimerization at the α-carbon of the ester group. The attachment of the third nitrogen was accomplished by the α-enamination of rac-13 using Bredereck's reagent.25 The desired intermediate rac-14 was obtained in almost 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio together with its epimer. The epimerization could be attributed to the tert-butoxy anion generated from Bredereck's reagent during the reaction. Variation of the solvent, temperature and reagent stoichiometry failed to tune the ratio between the two epimers. The reaction of rac-14 with sodium nitrite in cold acetic acid/water mixture provided oxime rac-15 as a mixture of the E/Z isomers in excellent yield. With everything set for the final lactamization, rac-15 was subjected to another pressurized palladium-catalysed hydrogenation to give diprotected trilactam rac-16. Even though the ring-closure was accelerated by acetic acid and the increased reaction temperature, it took four days for the reaction to be completed. The removal of the two PMP groups on rac-16 by ceric ammonium nitrate (CAN) was straightforward. However, the purification of the product was a challenge. Bearing three lactam groups within the structure, rac-1 exhibited higher affinity for water than for organic solvents. Therefore, extraction of the reaction crude during work-up with organic solvents did not result in the separation of rac-1 from the excess inorganic components. This issue was later solved by employing ion-exchange chromatography, where the uncharged product had a different retention time than the charged inorganic species. Further separation by HPLC yielded rac-1 as a white solid. Enantiopure 1 was also prepared. The low solubility of rac-1 in most solvents made it difficult to resolve the enantiomers by normal phase chiral chromatography. Instead, the more soluble precursor rac-16 was resolved on a preparative chiral column. Deprotection of (−)-16 followed by the same purification as for rac-1 resulted in (+)-1. The absolute configuration of (+)-1 was assigned as (+)-(SSS)-1 by comparing the experimental optical rotation and circular dichroism (CD) spectrum with corresponding quantum chemically calculated ones (see Scheme 1 and ESI†).
:1 ratio together with its epimer. The epimerization could be attributed to the tert-butoxy anion generated from Bredereck's reagent during the reaction. Variation of the solvent, temperature and reagent stoichiometry failed to tune the ratio between the two epimers. The reaction of rac-14 with sodium nitrite in cold acetic acid/water mixture provided oxime rac-15 as a mixture of the E/Z isomers in excellent yield. With everything set for the final lactamization, rac-15 was subjected to another pressurized palladium-catalysed hydrogenation to give diprotected trilactam rac-16. Even though the ring-closure was accelerated by acetic acid and the increased reaction temperature, it took four days for the reaction to be completed. The removal of the two PMP groups on rac-16 by ceric ammonium nitrate (CAN) was straightforward. However, the purification of the product was a challenge. Bearing three lactam groups within the structure, rac-1 exhibited higher affinity for water than for organic solvents. Therefore, extraction of the reaction crude during work-up with organic solvents did not result in the separation of rac-1 from the excess inorganic components. This issue was later solved by employing ion-exchange chromatography, where the uncharged product had a different retention time than the charged inorganic species. Further separation by HPLC yielded rac-1 as a white solid. Enantiopure 1 was also prepared. The low solubility of rac-1 in most solvents made it difficult to resolve the enantiomers by normal phase chiral chromatography. Instead, the more soluble precursor rac-16 was resolved on a preparative chiral column. Deprotection of (−)-16 followed by the same purification as for rac-1 resulted in (+)-1. The absolute configuration of (+)-1 was assigned as (+)-(SSS)-1 by comparing the experimental optical rotation and circular dichroism (CD) spectrum with corresponding quantum chemically calculated ones (see Scheme 1 and ESI†).
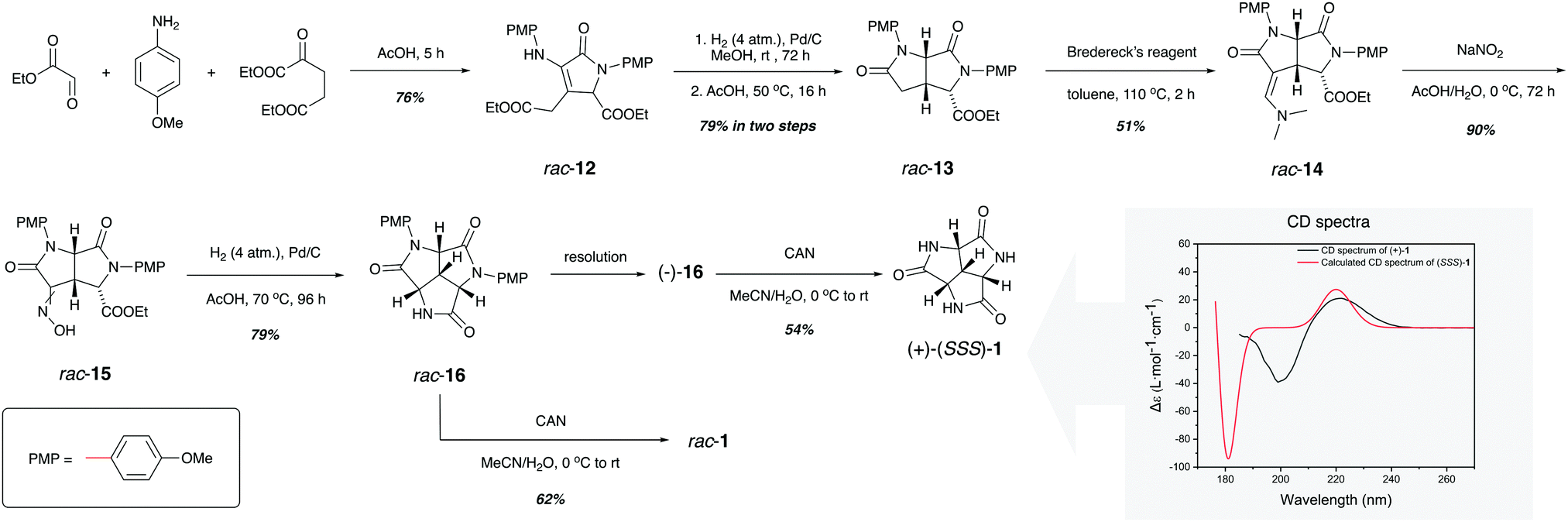 | ||
| Scheme 1 Synthesis of trilactam rac-1 and (+)-(SSS)-1 and the determination of the absolute configuration of (+)-1. | ||
Preliminary self-assembly studies in solution were carried out with and without guests for both rac-1 and (+)-(SSS)-1, respectively (see ESI†). However, no indication of host–guest complexation was observed in 1H NMR spectra. This could be attributed to the poor solubility of rac-1 and (+)-(SSS)-1 in less polar solvents such as chloroform and toluene, which can provide a benign environment for hydrogen bonding. For both rac-1 and (+)-(SSS)-1, the least polar solvent that could give sufficient solubility for NMR study was DMF-d7, which in general is too polar for hydrogen-bond based self-assembly.
Fortunately, we were more successful with solid-state self-assembly studies. Single crystals of rac-1-d3‡ were obtained from deuterium oxide by slow cooling and investigated by X-ray diffraction. As shown in Fig. 4a, the unit cell consists of two opposite-pointing stacks of trilactams. Each stack contains one (RRR)-1-d3 and one (SSS)-1-d3 that overlay along the c-axis via three pairs of weak C–H⋯O hydrogen bonds with the average C(H)⋯O distance of 3.55 Å. The two stacks are linked by a pair of glycine–glycine hydrogen bonds with the N(D)⋯O distance of 2.91 Å, which is within the range of typical hydrogen bond length for peptides.26 Closer inspection of the packing diagram reveals more interesting aspects. Viewing from [100], two layers of parallel zig-zag ribbons along [![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) 10] and [110] alternately stack along the c-axis (Fig. 4b and c). Each ribbon is assembled from alternating (RRR)-1-d3 and (SSS)-1-d3. Within each ribbon, the interaction between the adjacent trilactam alternates between glycine–glycine hydrogen bonds and lactam–lactam hydrogen bonds. Parallel ribbons of the same layer are cross-linked every other trilactam through another pair of lactam–lactam hydrogen bonds (Fig. 4c). This pattern could also be regarded as alternately stacked infinite layers of non-planar hexagonal tiling where alternating (RRR)-1-d3 and (SSS)-1-d3 assemble according to an unusual R46(24) graph set (Fig. 4d).27,28 Given that glycine–glycine interaction tends to give more stable assemblies compared to lactam–lactam interaction,17 it is interesting to note that in the present case the hexagonal pattern consists of two pairs of lactam–lactam hydrogen bonds and one pair of glycine–glycine hydrogen bonds. The energy demand for the formation of the energetic unfavourable pattern could be compensated by the efficient packing and the C–H⋯O hydrogen bonds between the layers.
10] and [110] alternately stack along the c-axis (Fig. 4b and c). Each ribbon is assembled from alternating (RRR)-1-d3 and (SSS)-1-d3. Within each ribbon, the interaction between the adjacent trilactam alternates between glycine–glycine hydrogen bonds and lactam–lactam hydrogen bonds. Parallel ribbons of the same layer are cross-linked every other trilactam through another pair of lactam–lactam hydrogen bonds (Fig. 4c). This pattern could also be regarded as alternately stacked infinite layers of non-planar hexagonal tiling where alternating (RRR)-1-d3 and (SSS)-1-d3 assemble according to an unusual R46(24) graph set (Fig. 4d).27,28 Given that glycine–glycine interaction tends to give more stable assemblies compared to lactam–lactam interaction,17 it is interesting to note that in the present case the hexagonal pattern consists of two pairs of lactam–lactam hydrogen bonds and one pair of glycine–glycine hydrogen bonds. The energy demand for the formation of the energetic unfavourable pattern could be compensated by the efficient packing and the C–H⋯O hydrogen bonds between the layers.
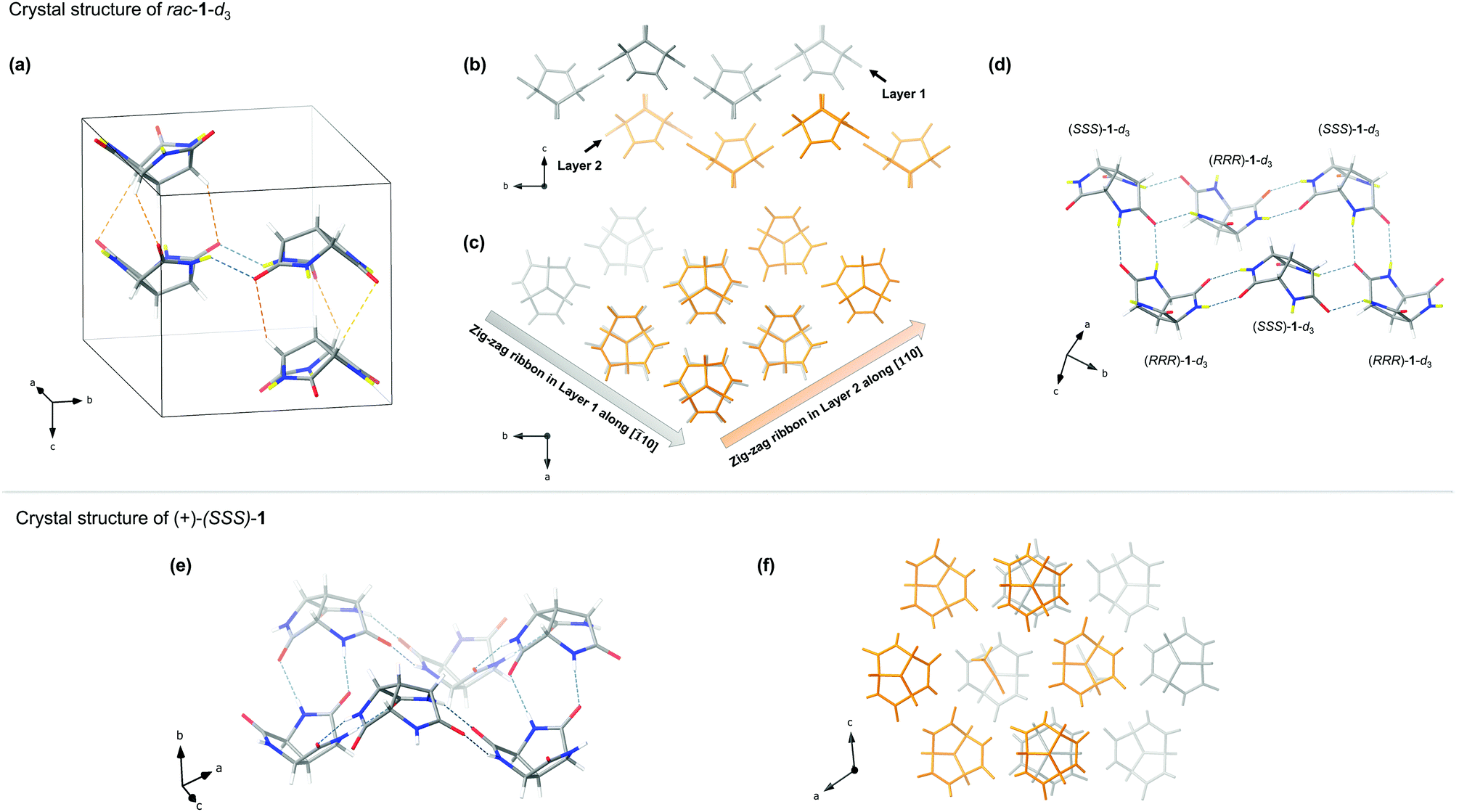 | ||
| Fig. 4 Crystal structures of rac-1-d3 and (+)-(SSS)-1. The hydrogen bonds are showed as dashed lines. (a) The unit cell of rac-1-d3; (b) The packing diagram of rac-1-d3 viewing from [100]; (c) The packing diagram of rac-1-d3 viewing from [001]; (d) The hexagonal tiling of rac-1-d3; (e) The hexagonal tiling of (+)-(SSS)-1; (f) The packing diagram of (+)-(SSS)-1 viewing from [010]. In (a), (d) and (e), grey = carbon atom; red = oxygen atom; blue = nitrogen atom; white = hydrogen atom; yellow = deuterium atom. | ||
Single crystals of (+)-(SSS)-1‡ were obtained from a water/acetonitrile mixture by layer diffusion. X-ray diffraction revealed the formation of infinite layers of a more regular hexagonal tiling which can be found in the crystals of similar compounds.10,17 In each hexagonal unit, six trilactams with alternating orientations are linked through glycine–lactam interaction according to the R66(27) graph set (Fig. 4e).27,28 The average length of the N(H)⋯O bonds is 2.87 Å, which is slightly shorter than those in racemic crystals. The layers stack along the b-axis in a repeating pattern where over the centre of each hexagonal unit there is a trilactam from the adjacent layer (Fig. 4f). The voids between the layers are occupied by acetonitrile molecules (Fig. 4f).
In summary, 51 years since it was first proposed as a synthetic target, the highly symmetric trilactam 1 was finally synthesized in both racemic and enantiopure form. Solid-state self-assembly of rac-1-d3 and (+)-(SSS)-1 were investigated by X-ray diffraction, which revealed two different patterns of hexagonal tiling through intermolecular hydrogen bonding. Further exploration into the self-assembly of rac-1 and (+)-(SSS)-1 in solution was hindered by their limited solubility in less polar aprotic solvents. Nevertheless, the successful synthesis of 1 opens new possibilities in host–guest chemistry, crystal engineering, chiral recognition, asymmetric catalysis as well as surface engineering. To be able to study the self-assembly of the trilactam in non-polar solvents, effort towards lipophilic derivatives of 1 is currently underway in our group.
K. W. thanks the Swedish Research Council (VR) and the Swedish Foundation for Strategic Research (Chemistry for life science program) for financial support. Y. L. acknowledges the Royal Physiographic Society of Lund for financial support and China Scholarship Council for PhD scholarship. We are grateful to Dr. Alexey Polukeev for helping to build the set-up for pressurized hydrogenation.
Conflicts of interest
The authors declare no competing financial interest.Notes and references
- S. N. V. R. H. Linnell, Hydrogen Bonding, Van Nostrand Reinhold Company, New York, 1971 Search PubMed.
- M. D. Joesten and L. J. Schaad, Hydrogen bonding, Marcel Dekker Inc, 1974 Search PubMed.
- G. A. Jeffrey, An Introduction to Hydrogen Bonding, Oxford University Press, 1997 Search PubMed.
- J. Rebek, Hydrogen-bonded Capsules: Molecular Behavior in Small Spaces, World Scientific, 2016 Search PubMed.
- G. R. Desiraju, Crystal Engineering: The Design of Organic Solids, Elsevier, 1989 Search PubMed.
- C. B. Aakeröy and K. R. Seddon, Chem. Soc. Rev., 1993, 22, 397–407 RSC.
- G. R. Desiraju, Angew. Chem., Int. Ed., 2007, 46, 8342–8356 CrossRef CAS PubMed.
- S. Saha, M. K. Mishra, C. M. Reddy and G. R. Desiraju, Acc. Chem. Res., 2018, 51, 2957–2967 CrossRef CAS PubMed.
- P. Baillargeon and Y. L. Dory, J. Am. Chem. Soc., 2008, 130, 5640–5641 CrossRef CAS PubMed.
- D. Beaudoin, F. Rominger and M. Mastalerz, Angew. Chem., Int. Ed., 2016, 55, 15599–15603 CrossRef CAS PubMed.
- S. Sartyoungkul, Y. Yakiyama and H. Sakurai, Asian J. Org. Chem., 2020, 9, 947–952 CrossRef CAS.
- C. Andreu, R. Beerli, N. R. Branda, M. Conn, J. de Mendoza, A. Galán, I. Huc, Y. Kato, M. Tymoschenko, C. Valdez, E. Wintner, R. Wyler and J. Rebek, Pure Appl. Chem., 1993, 65, 2313–2318 CrossRef CAS.
- W. L. Mock, J. Org. Chem., 1971, 36, 3613–3614 CrossRef CAS.
- C. Moberg, Angew. Chem., Int. Ed., 1998, 37, 248–268 CrossRef CAS PubMed.
- S. E. Gibson and M. P. Castaldi, Chem. Commun., 2006, 3045–3062 RSC.
- C. Moberg, Angew. Chem., Int. Ed., 2006, 45, 4721–4723 CrossRef CAS PubMed.
- N. Branda, PhD thesis, Massachusetts Institute of Technology, 1994.
- The estimation of cavity volumes was performed by MSRoll with the probe radius 1.0 Å, 1.7 Å and 3.7 Å, respectively.
- J. A. Theobald, N. S. Oxtoby, M. A. Phillips, N. R. Champness and P. H. Beton, Nature, 2003, 424, 1029–1031 CrossRef CAS PubMed.
- J. Teyssandier, S. D. Feyter and K. S. Mali, Chem. Commun., 2016, 52, 11465–11487 RSC.
- T. Jasper-Tönnies, M. Gruber, S. Ulrich, R. Herges and R. Berndt, Angew. Chem., Int. Ed., 2020, 59, 7008–7017 CrossRef PubMed.
- Personal communication with Professor Mock by e-mail on 26 September 2003.
- At the time a PhD student in Julius Rebek's group.
- X. del Corte, A. Maestro, J. Vicario, E. Martinez de Marigorta and F. Palacios, Org. Lett., 2018, 20, 317–320 CrossRef CAS PubMed.
- M. S'kof, J. Svete, M. Kmetič, S. Golič-Grdadolnik and B. Stanovnik, Eur. J. Org. Chem., 1999, 1581–1584 CrossRef.
- I. L. Karle, J. Mol. Struct., 1999, 474, 103–112 CrossRef CAS.
- M. C. Etter, Acc. Chem. Res., 1990, 23, 120–126 CrossRef CAS.
- J. Bernstein, R. E. Davis, L. Shimoni and N.-L. Chang, Angew. Chem., Int. Ed. Engl., 1995, 34, 1555–1573 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d2cc00345g |
| ‡ Crystal data for rac-1-d3: C7H4D3N3O3, M = 184.17, monoclinic, a = 7.2905(7), b = 11.3194(9), c = 9.0781(10) Å, U = 695.89(13) Å3, T = 293(2) K, space group P21/c (no. 14), Z = 4, 4010 reflections collected, 1622 unique (Rint = 0.0249). The final wR2 was 0.1184 (all data). Crystal data for (+)-(SSS)-1: [2(C7H7N3O3), C2H3N], M = 403.36, monoclinic, a = 10.2416(18), b = 9.0837(12), c = 10.5564(16) Å, U = 873.1(3) Å3, T = 293(2) K, space group P21 (no. 4), Z = 2, 10598 reflections collected, 10598 unique. The final wR2 was 0.2876 (all data). |
| This journal is © The Royal Society of Chemistry 2022 |