
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA guanidino-γ-cyclodextrin superdimer generates a twin receptor for phosphate dimers assembled by anti-electrostatic hydrogen bonds†
Emmanuel
Saridakis
 ,
Eleni-Marina
Kasimati
,
Konstantina
Yannakopoulou
* and
Irene M.
Mavridis
*
,
Eleni-Marina
Kasimati
,
Konstantina
Yannakopoulou
* and
Irene M.
Mavridis
*
Institute of Nanoscience and Nanotechnology, National Center for Scientific Research “Demokritos”, Patr. Grigoriou E’ & 27 Neapoleos str, Aghia Paraskevi Attikis 15341, Greece. E-mail: k.yannakopoulou@inn.demokritos.gr; e.mavridis@inn.demokritos.gr
First published on 1st April 2022
Abstract
Octakis-6-guadinidino-γ-cyclodextrin (gguan) hydrochloride in the presence of phosphates crystallises from aqueous solution in the unprecedented form of a superdimer (dimer-within-a-dimer). The self-assembly exposes four circular octa-guanidinium regions that bind and stabilise discrete H-bonded phosphate anion dimers. The small (∼2 nm) gguan-phosphate assembly is preorganised and stable in aqueous solution, as demonstrated by DLS and NMR experiments.
Supramolecular structures formed via self-assembly of suitable components are of immense importance in nature. Research to create novel, interesting artificial supramolecular architectures and materials by the combination of binding motifs based on multiple H-bonds and electrostatic/hydrophobic interactions between synthetic components, either in non-polar or polar environments, is very actively pursued.1,2 Cyclodextrins (α, β and γCDs) are macrocyclic carbohydrate oligomers that can entrap a vast variety of compounds via non-covalent bonds in aqueous solution to form supramolecular inclusion complexes with multiple applications.3 The CD structure is lined with primary and secondary hydroxy (OH) groups. The latter establish a strong intramolecular H-bonding network between vicinal glucopyranose units, crucial for the rigidity of the macrocycle, but they can also form well-organised H-bonds with the secondary OHs of another CD and create dimers, usually in the presence of a suitably sized guest molecule, that aligns the two hosts. The dimer constitutes a frequent motif encountered in the crystalline state4,5 but also in solution.6,7
Regioselective modifications of OHs are employed with the ultimate goal of enhancing the inclusion capability of CDs via the establishment of additional interactions with suitable guests. For instance, attachment of cationic8 or anionic groups9 on the primary CD side has been instrumental in capturing very efficiently anionic10 or cationic11 guests, respectively, and in coordinating metal cations9 for important bio-applications. Charged groups also endow the CD hosts with new properties, beyond inclusion: positively charged CDs and CD-based systems function as cell-penetrating and DNA-transfection agents.8,12 Being water soluble oligomeric salts, none of the above positively charged CDs or similar derivatives8 reported in the literature have been crystallised, to our knowledge. The exact structure of such hosts and their interactions with counter ionic substrates has thus been elusive.
Herein we report on the molecular structure of octakis(6-guanidino-6-deoxy)-γCD (gguan) that has been crystallised in the presence of phosphates in the unprecedented form of a superdimer, namely an inner core dimer enclosed within an outer dimer, exhibiting interesting molecular and supramolecular symmetries. A striking element of the structure is the presence of two phosphate dimers assembled via anti-electrostatic H-bonds13 which are embedded within the core dimer's guanidinium group peripheries and serve as stabilising moieties that are also crucial for crystallisation. Additional phosphate pairs held together through numerous interactions, contribute to the growth of the crystal lattice. DLS and NMR studies show that the superdimer assembly preexists in solution and confirm the critical role of phosphates in organizing the structure. To our knowledge, such a 3-level assembly (phosphate pairs-core dimer-outer dimer) has not been reported before.
Single crystals were obtained from an aqueous solution of gguan·8HCl by hanging-drop vapor diffusion using 2-methyl-2,4-pentanediol (MPD) and monobasic ammonium phosphate in Tris buffer of pH 8.5 (see ESI†). Such conditions, from the protein crystallisation toolbox, are very uncommon for crystallisation of CDs. An improvement in diffraction resolution was achieved by introducing the heterogeneous nucleation-inducing agent Bioglass under metastable conditions.14
The structure at 1.10 Å resolution (Table S1, ESI†) comprises two crystallographically independent gguan molecules, A and B (Fig. 1a) and 4.6 phosphate counter-ions distributed over 7 sites, three of full and four of partial occupancies. Moreover, numerous disordered water molecules and one ammonium ion are located inside and around the two monomers, A and B, which are nearly perpendicular to each other (88°). By rotation about the b axis, the oblong superdimer ABAsymBsym is generated (Fig. 1b and c). A, Asym form a core dimer, which is the guest for the B, Bsym outer dimer. The stability of the whole assembly is maintained by numerous H-bonds. Interestingly, the eight most prominent H-bonds are established between outward-pointing guanidinium nitrogen atoms of guests A and Asym and secondary OH groups of B and Bsym (Fig. S2, ESI†). The γCD moieties of both A and B are of a rather undistorted geometry (Table S2, ESI†) and exhibit intramolecular H-bonds between the secondary OH groups that contribute to the rigidity of the torus, as in α- and βCDs.5 The planar guanidinium groups adopt the (+)-gauche orientation that results in one nitrogen atom (N2) pointing inwards over the cavity and the other (N3) outwards (Fig. 2a and Fig. S1, S3, Table S3, ESI†). The former generates a cyclic belt in the internal periphery of the primary sides of A and B, where pairs of phosphates are embedded differently in the two monomers (Fig. 2). Three additional phosphate anions connect the supermolecules in the lattice.
 | ||
| Fig. 1 (a) Face ac in front; the asymmetric unit, A and B molecules; (b) superdimer AAsymBBsym, viewed as in (a), displaying a symmetric shape. (c) Face bc in front; AAsymBBsym along the a axis; in this orientation the shape is not symmetric. Generated by PyMOL.15 | ||
 | ||
| Fig. 2 (a) The archetypal phosphate dimer P2–P3 is assembled by AEHBs (2.45 and 2.95 Å) and is confined within the guanidinium cation belt at the primary sides of A and Asym, with mutually stabilising electrostatic H-bonds. Shown also is the external phosphate P1, H-bonding to the guanidinium cation of G1. (b) Phosphates P4 and P7 of the B and Bsym primary sides do not form a typical anion–anion dimer (shortest distance between O atoms of P4 and P7, 3.6 Å). (c) The external P1–P1sym phosphate dimer is found in a pocket between superdimers enveloped by guanidinium cations and other H-donors. | ||
Monomers A and B exhibit a pseudo 2-fold symmetry with respect to their molecular axis. The pseudo axis defines two sets relating glucopyranose residues G2, G3, G4, G5 to G6, G7, G8, G1, respectively (notation in Fig. 2 and Fig. S1, ESI†). This is more visible in the planar guanidinium group, exhibiting different orientations between the two sets of nearly parallel guanidinium planes (shortest interplanar distances ∼3.4 Å, Fig. 2, and Fig. S3, Table S3, ESI†). Molecules A and Asym of the core dimer are coaxial, stabilised by eight very strong intermolecular O–H⋯O H–bonds, (Table S4, ESI†). It is noteworthy that four of these eight H-bonds are identical to the other four (Table S4 and Fig. S4, ESI†). This is due to the location of the atoms OA41 and CA61 in A and their symmetry equivalent atoms in Asym, which define a plane containing the 2-fold b axis relating the two monomers (Fig. S4, ESI†).
Within the primary sides of A, Asym, two phosphate anions are enclosed, P2 and P3, at O⋯O distances of 2.45 and 2.95 Å (Fig. 2a). At the pH of the crystallisation conditions, the phosphate anions co-exist as a mixture16 of di- and mono-anions, namely ∼85% HPO42− and ∼15% H2PO4− and the strongly basic guanidino groups are protonated (vide infra). Given the predominance of the doubly charged species we postulate that two HPO42− anions assemble as a P2–P3 dimer by anti-electrostatic H-bonds (AEHBs)13,17 and are further stabilised via electrostatic H-bonds by the surrounding, positively charged, inward-pointing N2 guanidinium atoms (Fig. 2a and Table S5, ESI†), namely residues of the G2–G5 set on the one side of the P2–P3 dimer and those of the G6–G1 set on the other (range of N⋯O distances 2.6–3.1 Å).
The guanidinium groups of the B monomers (Fig. 2b) are also segregated into two sets of four and the inward-pointing N2 atoms of each set of residues establish electrostatic H-bonds separately along each side of the two confined phosphate anions P4 and P7 (Fig. 2b). However, the two phosphate groups do not form, in terms of their mutual distance (3.6 Å) and relative positions, an archetypal anion–anion dimer. P7 is more exposed than P4, as it extends slightly out of the guanidium belt, actually coming closer to the external phosphate P6 (distance 3.1 Å) than to the internal P4 (Fig. 2b and Table S5, ESI†), thus forming a linear trimer association.18
The remaining three phosphates P1, P5, P6 are located at interstitial positions between superdimers (Figs. S5, S6, ESI†) and contribute to their connection by H-bonding to outward pointing N3 and OH groups, as well as indirectly through the numerous water molecules. It is noteworthy that phosphate P1 forms a dimer with its symmetry equivalent P1sym. This is established directly by one anion-anion H-bond (O⋯O distance of 2.5 Å) and indirectly by two ion pair bridges with symmetry related ammonium cations (Fig. 2c). Located in a pocket confined between two neighbouring CD superdimers, each P1 phosphate establishes also direct H-bonds with the guanidinium atoms N2 and N3 of residue G1 of one of the two superdimers. Thus, P1–P1sym is another type of phosphate dimer stabilised by H-bonding interactions in a “broader sense”, as observed in other cases of anion-anion dimers.17,18
Summarising, phosphates P2 and P3, of full occupancy, form a very strong archetypal dimer assembled by AEHBs inside the positively charged belt of the core-dimer AAsym, which constitutes a twin receptor for the phosphate dimers. In contrast, phosphates P4 and P7, of almost half occupancy and more mobile (probably an average of two or more disordered positions), show a “dissociating” or breaking up phosphate dimer,13 still stabilised by H-bonding with the guanidinium groups of B,Bsym. Finally, the packing and the outward pointing N3 atoms allow for the appropriate environment to stabilise a P1–P1sym dimer by a variety of H-bonds with positively charged and neutral H-donor groups.
DLS and NMR experiments were performed using the same conditions as those employed to obtain the single crystals, to investigate if the gguan-phosphate assembly is an effect of the solid state or it is the result of a stable pre-organisation of the molecules in aqueous solution. Therefore, 8 mM gguan·8HCl solutions prepared in Tris buffer pH 8.5 were monitored over time with and without NH4H2PO4. Borate buffer solutions of pH 8.5 in H2O and D2O were also monitored in parallel by DLS and 1H/31P NMR spectra, at the same time points as the Tris samples.
The DLS results in Tris buffer with (Fig. 3 A–D) and without (Fig. 3A′–D′) NH4H2PO4 are presented as plots of intensity vs. % size distribution. It is clearly observed that the large and polydisperse species of gguan (Fig. 3A and A′) within 1 day following addition of NH4H2PO4, are transformed into still polydisperse but smaller, nanosized particles (Fig. 3B). Impressively, the species reach nearly uniform ∼2 nm populations after 3 days (Fig. 3C), and this is approximately the time (2–3 days) when the crystals appear during crystallisation. After 6 days the stability of the small particles seems somewhat disturbed (Fig. 3D). The results after 3 days are identical in both the intensity and number vs. % size distribution diagrams (Fig. S7aF, ESI†), indicating the presence of only very small (∼2 nm) and quite uniform particles. These observations are also valid for the sequence of graphs in borate buffer solution (Fig. S7b, ESI†), where the nanoparticles reach nearly the same uniformity and small size after 3 days (Fig. S7bF, ESI†), as in the Tris series. In contrast, in Tris without NH4H2PO4, gguan particles appear in various sizes over the entire size range and they are not particularly affected with time, lacking any appreciable tendency to form only small-sized species (Fig. 3B′ and C′). It should be noted that visible crystals appear in Tris buffer with NH4H2PO4 and MPD after 2–3 days, whereas none of the trials afforded crystals in the absence of NH4H2PO4. The DLS results therefore suggest that both Tris and borate buffer solutions at pH 8.5 elicit almost the same gguan behaviour, but what seems to be a decisive factor to organising gguan in small (∼2 nm) and uniform populations is the presence of NH4H2PO4. The fact that the small DLS aggregates present after 3 days are destabilised after 6 days suggests that the assembly is metastable. Therefore, it is interesting that it has been possible to “capture” it using screens and approaches suitable for protein crystallisation. Moreover, using (NH4)2SO4 instead of NH4H2PO4 gave mixed populations after 1, 3 and 6 days in the same DLS conditions (Fig. S8, ESI†) and yielded no crystals in crystallisation trials, thus attesting to the organising role of the phosphates.
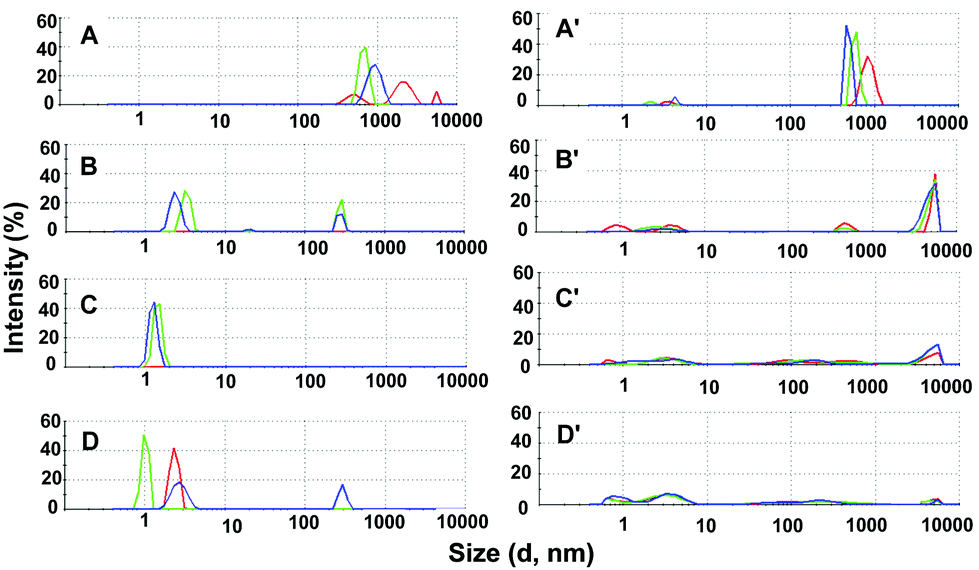 | ||
| Fig. 3 DLS experiments in Tris buffer (100 mM, pH 8.5) shown as % size distribution by intensity: (A) gguan·8HCl (8 mM) right after dissolution, (B) after 1 day with NH4H2PO4 (200 mM), (C) after 3 days, and (D) after 6 days. Diagrams A′–D′ are of measurements at the same time-points, but without NH4H2PO4. | ||
In support of the above, 1H NMR experiments with gguan in deuterated borate buffer solution (Fig. S9A, ESI†) show that addition of NH4H2PO4 affects visibly the chemical shifts of the protons H6, H6′ only (Fig. S9B, ESI†), suggesting establishment of stabilising phosphate-guanidinium interactions that result in an unmodified 1H NMR spectrum after 3 days (Fig. S9C, ESI†). On the other hand, repeated dialyses of the gguan in Tris with NH4H2PO4 DLS sample and acquisition of the 1H NMR spectrum in D2O (Fig. S9D, ESI†) reveals high similarity to the 1H spectrum after 3 days (Fig. S9C, ESI†) and the persistent presence of phosphate in the 31P spectrum (Fig. S9D1, D2, ESI†), indicating sustained phosphate–guanidinium association.
Additionally, pKa determination was performed with 13C NMR titrations of a gguan solution either in H2O or in H2O/1 M KCl19 and the signals of guanidino C7 (∼157 ppm), and CD core C6 and C5 were monitored (numbering in Fig. S9, ESI†). A small signal at ∼162 ppm that corresponds to gguan carbonate emerged at pH ∼10.5 and increased both with time and with pH, indicating the absorption of atmospheric CO2. Fitting of the titration data in H2O/1 M KCl to the appropriate equation (Δδ13Cvs. pH, ESI†) afforded unreasonable pKa values or values around 14 (Fig. S10, ESI†). It is worth noting that at pH < 7, 1H NMR signals at 6.81 ppm (NH) and 6.24 ppm (–C(![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) NH)NH3+) are clearly observed (Fig. S11, ESI†), despite the presaturation irradiation applied, reflecting the very high basicity of the guanidino groups. The above results suggest that the step-wise deprotonation of the guanidino groups ranges from pH 10.5 to around 14, which is expected in this class of per-functionalised positively charged CDs20 and that gguan has very high global pKa. It has to be noted that recent publications place the pKa value of the arginine side chain at 13.8,21 as opposed to the usually reported value of ∼12. In the case of gguan, the pH of the crystallization medium after addition of NH4H2PO4 becomes ∼8.0, and thus all guanidino groups are completely protonated.
NH)NH3+) are clearly observed (Fig. S11, ESI†), despite the presaturation irradiation applied, reflecting the very high basicity of the guanidino groups. The above results suggest that the step-wise deprotonation of the guanidino groups ranges from pH 10.5 to around 14, which is expected in this class of per-functionalised positively charged CDs20 and that gguan has very high global pKa. It has to be noted that recent publications place the pKa value of the arginine side chain at 13.8,21 as opposed to the usually reported value of ∼12. In the case of gguan, the pH of the crystallization medium after addition of NH4H2PO4 becomes ∼8.0, and thus all guanidino groups are completely protonated.
The DLS and NMR studies demonstrate that a stable, very small-sized gguan-phosphate assembly pre-exists in solution. Regarding crystallisation, it has been proposed that the Hofmeister sequence of anions, as applied to proteins, is analogous to the salting-out series for small molecules.22 Therefore, the use of kosmotropic phosphate in crystallisation can be considered as a means of stabilising both the gguan molecule and its tetramer in solution (and possibly cooperating with MPD to salting it out). Therefore, we propose that the coexistence of NH4H2PO4 and gguan at pH ∼8 triggers multiple phases of organisation in aqueous solution. A working model could be that the phosphate anions, presumably HPO42− species,16 are organised as dimers in the ideal environment (in terms of charge and geometry) of the primary sides of the macrocycle. This apparently stabilises the conformation of the guanidinium groups surrounding the phosphates and facilitates the self-assembly of gguan first to core dimers and subsequently to tetrameric superdimers in solution. Finally, the phosphates located outside the gguan cavities establish direct H-bonds with outward pointing N3 groups of the guanidinium cations, mediating the development of the intermolecular bonds that build the crystal lattice, whereas (NH4)2SO4 is unsuccessful. The present crystallisation approach could be used to crystallise similar, highly water-soluble charged macrocycles.
Designed guanidinium receptors for phosphate anion binding have been based on H-bonding interactions between opposite charges23 following mostly an enthalpy driven process with positive entropy changes in polar/protic environments.24 These and other studies are based, however, on the highly directional binding of the Y-shaped guanidino groups to phosphate oxo-anions via both ion-pairing and H-bonding, as encountered in natural systems. Here, however, we observe phosphate anion dimers inside each gguan periphery that compel the guanidino groups to rotate perpendicularly over the cavity to form multiple H-bonds and neutralize the charges, thus creating a new guanidino–phosphate binding motif. Recently, there has been a strong interest in receptors25 that form complexes with anion–anion dimers assembled by AEHBs offering an appropriate stabilising environment. In the present study we demonstrate that gguan and its quadruple superdimer assembly serve perfectly the requirements for such receptors, especially for “highly anti-electrostatic”13 phosphate dimers, stable in aqueous solutions.
E. S.: investigation, methodology, writing-review-editing; E. M. K.: investigation, methodology, writing-original draft; K. Y.: conceptualisation, investigation, writing-review-editing, supervision, resources; I. M. M: conceptualization, investigation, methodology, writing-review-editing.
Funding by (i) the National Research Infrastructures on Integrated Structural Biology, Drug Screening Efforts and Drug Target Functional Characterization-INSPIRED project, MIS 5002550 and (ii) the Operational Programme “Competitiveness, Entrepreneurship and Innovation” (NSRF 2014-2020), KRIPIS II project, MIS 5002567, co-financed by Greece and the European Union, is gratefully acknowledged.
Conflicts of interest
There are no conflicts of interest to declare.Notes and references
- Hydrogen Bonded Supramolecular Structures, ed. Z.-T. Li and L.-Z. Wu, SpringerBerlin, Heidelberg, Berlin, 2015 Search PubMed.
- J. N. Smith, C. Ennis and N. T. Lucas, Chem. Sci., 2021, 11858 RSC.
- G. Krini, Chem. Rev., 2014, 114(21), 10940–10975 CrossRef PubMed.
- K. Harata, Chem. Rev., 1998, 98, 1803–1827 CrossRef CAS PubMed.
- S. Makedonopoulou and I. M. Mavridis, Acta Crystallogr., 2000, B56, 322–331 CAS.
- G. Wenz, B.-H. Han and A. Müller, Chem. Rev., 2006, 106, 782–817 CrossRef CAS PubMed.
- N. Mourtzis, G. Cordoyiannis, G. Nounesis and K. Yannakopoulou, Supramol. Chem., 2003, 15, 639–649 CrossRef CAS.
- C. O. Mellet, J. M. G. Fernández and J. M. Benito, Chem. Soc. Rev., 2011, 40, 1586–1608 RSC.
- I. M. Mavridis and K. Yannakopoulou, Int. J. Pharm., 2015, 492, 275–290 CrossRef CAS PubMed.
- V. Rodriguez-Ruiz, A. Maksimenko, G. Salzano, M. Lampropoulou, Y. G. Lazarou, V. Agostoni, R. Gref and K. Yannakopoulou, Sci. Rep., 2017, 7, 8353 CrossRef PubMed.
- A. Bom, M. Bradley, K. Cameron, J. K. Clark, J. van Egmond, H. Feilden, E. J. MacLean, A. W. Muir, R. Palin, D. C. Rees and M.-Q. Zhang, Angew. Chem., Int. Ed., 2002, 41, 265–270 CrossRef CAS.
- N. Mourtzis, M. Paravatou, I. M. Mavridis, M. L. Roberts and K. Yannakopoulou, Chem. – Eur. J., 2008, 14, 4188–4200 CrossRef CAS PubMed.
- W. Zhao, A. H. Flood and N. G. White, Chem. Soc. Rev., 2020, 49, 7893–7906 RSC and references therein.
- N. E. Chayen, E. Saridakis and R. P. Sear, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 597–601 CrossRef CAS PubMed.
- W. L. DeLano, The PyMOL Molecular Graphics System, DeLano Scientific LLC, 2002, https://www.pymol.org Search PubMed.
- M. Śmiechowski, E. Gojło and J. Stangret, J. Phys. Chem. B, 2009, 113, 7650–7661 CrossRef PubMed.
- W. Zhao, J. Tropp, B. Qiao, M. Pink, J. D. Azoulay and A. H. Flood, J. Am. Chem. Soc., 2020, 142, 2579–2591 CrossRef CAS PubMed.
- A. Rajbanshi, S. Wan and R. Custelcean, Cryst. Growth Des., 2013, 13, 2233–2237 CrossRef CAS.
- K. Popov, H. Rönkkömäki and L. H. J. Lajunen, Pure Appl. Chem., 2006, 78, 663–675 CrossRef CAS.
- G. Wenz, C. Strassnig, C. Thiele, A. Engelke, B. Morgenstern and K. Hegetschweiler, Chem. – Eur. J., 2008, 14, 7202–7211 CrossRef CAS PubMed.
- C. A. Fitch, G. Platzer, M. Okon, G.-M. E. B. and L. P. McIntosh, Protein Sci., 2015, 24, 752–761 CrossRef CAS PubMed.
- A. M. Hyde, S. L. Zultanski, J. H. Waldman, Y.-L. Zhong, M. Shevlin and F. Peng, Org. Process Res. Dev., 2017, 21, 1355–1370 CrossRef CAS.
- P. Blondeau, M. Segura, R. Pérez-Fernández and J. de Mendoza, Chem. Soc. Rev., 2007, 36, 198–210 RSC and references therein.
- V. D. Jadhav, E. Herdtweck and F. P. Schmidtchen, Chem. – Eur. J., 2008, 14, 6098–6107 CrossRef CAS PubMed and references cited.
- Q. He, P. Tu and J. Sessler, Chem, 2018, 4, 46–93 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2133070. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2cc00323f |
| This journal is © The Royal Society of Chemistry 2022 |