
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceOrganotelluroxane molecular clusters assembled via Te⋯X− (X = Cl−, Br−) chalcogen bonding anion template interactions†
Andrew
Docker
*a,
Antonio J.
Martínez Martínez
 b,
Heike
Kuhn
a and
Paul D.
Beer
*a
b,
Heike
Kuhn
a and
Paul D.
Beer
*a
aChemistry Research Laboratory, Department of Chemistry, University of Oxford, Mansfield Road, Oxford, OX1 3TA, UK. E-mail: andrew.docker@chem.ox.ac.uk; paul.beer@chem.ox.ac.uk
bSupramolecular Organometallic and Main Group Chemistry Laboratory, CIQSO-Center for Research in Sustainable Chemistry and Department of Chemistry, University of Huelva, Campus El Carmen, ES-21007 Huelva, Spain
First published on 11th February 2022
Abstract
The synthesis and characterisation of two novel molecular organotelluroxane clusters, comprising of an inorganic Te8O6X4 (X = Cl, Br) core structure are described. The integration of highly electron withdrawing 3,5-bis-trifluoromethylphenyl groups to the constituent Te(IV) centres is determined to be crucial in the chalcogen bonding (ChB) halide template directed assembly. Characterised by multi-nuclear 1H, 125Te, 19F NMR, UV-Vis, IR spectroscopies and X-ray crystal structure analysis, the discrete molecular clusters exhibit excellent organic solvent solubility and remarkable chemical stability. Furthermore, preliminary fluorescence investigations reveal the telluroxanes exhibit aggregation induced emission (AIE) behaviour in organic aqueous solvent mixtures.
In the construction of large, highly ordered macromolecular assemblies, the use of metal template-directed structural components have featured heavily.1–3 In particular, transition metal cations possessing predictable coordination geometries and thermodynamically stable complexes have been successfully exploited in generating elaborate and complex molecular topologies.1,4–7 However, recent years have witnessed exotic sigma-hole based non-covalent interactions, such as halogen bonding (XB) and chalcogen bonding (ChB) being exploited for recognition-derived spontaneous assembly.8–13 In this context, seminal work by Vargas-Baca and others have elegantly demonstrated that rational and considered incorporation of ChB donor–acceptor arrays in complementary molecular subunits can provide access to a diverse library of impressive, functional supramolecular assemblies.14–22 Organotelluroxanes, containing highly polar Te–O bonds, have shown enormous promise in this regard,23,24 frequently displaying intriguing structural behaviour forming oligomeric, polymeric and macrocyclic networks dictated by tellurium centred electrophilic interactions.25–28 Our own research endeavours have focused on the development of ChB donors and related sigma-hole interactions as anion recognition motifs which through the strategic selection of key structural and electronic factors tune anion binding potency and selectivity.29–32 Recently, we and others have demonstrated the Lewis acidity of tellurium-based sigma-hole donors is highly sensitive to local electronic environments and can be dramatically enhanced by integration of electron-withdrawing groups, especially when directly bound to the chalcogen atom.33–36 Motivated by these observations, we sought to establish whether this strategy of increasing Te-based sigma-hole donor potency through electronic polarisation could be exploited in the directed assembly of telluroxane architectures. Herein, we explore the synthesis of new telluroxanes via hydrolysis of diaryl tellurium dihalides (Ar2TeX2, X = Cl, Br), bearing highly electron withdrawing 3,5-bis-trifluoromethylphenyl (Ar) substituents. In the presence of a halide templating agent, a molecular Ar16Te8O6X4 cluster is spontaneously formed (Fig. 1). Characterised by a suite of spectroscopic techniques and X-ray diffraction structural analysis, the clusters are comprised of an Te8O6X4 core assembled through Te⋯X− (X− = Cl−, Br−) ChB-anion interactions. Notably, the telluroxane clusters exhibit highly desirable physical properties, including organic solvent solubility and chemical stability. Furthermore, preliminary photophysical investigations reveal the clusters exhibit aggregation induced emission (AIE) behaviour, becoming highly fluorescent upon the formation of hydrophobically driven aggregates.
 | ||
| Fig. 1 Chemical structure of Ar16Te8O6X4 clusters. | ||
The main method employed for the preparation of organotelluroxane compounds is the controlled hydrolysis of their respective diorgano tellurium(IV) dihalide (Ar2TeX2).24 With the objective of increasing ChB sigma-hole donor potency, the integration of highly electron withdrawing bis-trifluoromethylphenyl groups as inductively activating aryl appendages to the chalcogen centre was undertaken. Access to the requisite Ar2TeX2 (X = Cl, Br) species was explored through two routes, A and B, summarised in Scheme 1. Route A involved the treatment of 1,3-bis-trifluoromethylbromo benzene with n-BuLi in anhydrous Et2O at −78 °C, affording the corresponding organolithium species via a lithium halogen exchange reaction. Subsequent addition of an anhydrous Et2O TeCl4 or TeBr4 suspension gave the respective Ar2TeX2 species. Hydrolysis was achieved by carefully controlled addition of an NH4X(aq) solution to the crude reaction mixtures. Route B required the of isolation of diaryl telluride 3, which was obtained by treatment of diaryl ditelluride 2 with a 1 M Br2 CH2Cl2 solution to afford the corresponding organotellurium bromide, which was reacted immediately with a freshly generated THF solution of 1,3-bis-trifluoromethyl-phenylmagnesium bromide. Oxidative halogenation of 3, via addition of a Cl2 or Br2 CH2Cl2 solution, afforded the Ar2TeX2 species which was subjected to an analogous hydrolysis procedure with NH4X(aq). Identical aqueous work up procedures were undertaken for each of the crude reaction mixtures, in which TLC analysis revealed the formation of one major species. Isolation of these species from the hydrolysis (either Route A or B) of the diorgano tellurium dichloride or dibromide by column chromatography afforded the corresponding products, 1·Cl and 1·Br, as highly organic solvent soluble colourless solids. It is noteworthy that analogous reaction conditions in which the hydrolysis of the Ar2TeX2 species was attempted in the absence of NH4X, gave intractable mixtures of highly insoluble products, and no detectable amounts of the telluroxane cluster products. Furthermore, while routes A and B gave comparable yields of 1·Cl and 1·Br (see ESI†), attempts to conduct similar hydrolysis reactions of Ar2TeI2 (accessible by Route B) gave no isolable product. Inspection of the 1H, 125Te and 19F NMR spectra in acetone-d6 of the isolated products 1·Cl and 1·Br, revealed highly similar and relatively simple spectra (Fig. S18a, ESI†). The 1H NMR spectrum showed only two signals, in which integration determined a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 ratio, consistent with those belonging to a bis-trifluoromethylphenyl group. The 125Te spectra similarly indicated the presence of one resolved signal at ca. 820 ppm, consistent with the typical chemical shifts observed for a tellurane (O–Ar2Te–O) and similarly the 19F NMR spectra also indicated one resonance corresponding to the trifluoromethyl groups. Importantly, it was also observed that the NMR signals for 1·Cl or 1·Br were concentration independent (1–50 mM), implying the absence of non-covalent interactions between the isolated species in solution phase. The UV-Vis spectra of 1·Cl and 1·Br exhibited principal absorptions at 262 and 300 nm (Fig. S18b, ESI†), whereas in the IR, a complex profile of the phenyl ring and intense absorptions from the CF3 groups in the region 1350–1000 cm−1 were observed. Notably, another distinct sharp absorption at 679 cm−1 consistent with the characteristic tellurane (Te–O) stretch was also seen for both 1·Cl and 1·Br (Fig. S18c and S15, ESI†).37 Interestingly, at wavenumbers larger than 1350 cm−1, 1·Cl and 1·Br demonstrated excellent IR transparency, importantly indicating the absence of hydroxyl functionalities (Fig. 18c, ESI†).38 ESI-MS analysis of 1·Cl and 1·Br revealed the presence of several telluroxane cations including; [Ar2TeO]+ (m/z = 572.9), [(Ar2Te)2O2]+ (m/z = 1140.8), [(Ar2Te)2O2]+ (m/z = 1140.8) and [(Ar2Te)3O3]+ (m/z = 1712.9), which are presumably the result of autoionization under MS conditions (Fig. S16, ESI†).28 Determined melting points of 1·Cl and 1·Br (47 °C and 49 °C respectively) are in stark contrast with the vast majority of reported telluroxane structures, which typically exhibit high melting points (>100 °C).26,28,38,39 The combined spectroscopic evidence, such as, resolved concentration independent NMR signals and physical properties including low sharp melting points and excellent solubility in a wide range of non-polar solvents (e.g. dichloromethane, hexane, toluene, Fig. S4–S6, ESI†) all suggests that both 1·Cl and 1·Br are discrete molecular entities. It is noteworthy that this is contrary to the vast majority of telluroxanes reported to date, which are invariably oligomeric or polymeric structures of largely ionic character.
:2 ratio, consistent with those belonging to a bis-trifluoromethylphenyl group. The 125Te spectra similarly indicated the presence of one resolved signal at ca. 820 ppm, consistent with the typical chemical shifts observed for a tellurane (O–Ar2Te–O) and similarly the 19F NMR spectra also indicated one resonance corresponding to the trifluoromethyl groups. Importantly, it was also observed that the NMR signals for 1·Cl or 1·Br were concentration independent (1–50 mM), implying the absence of non-covalent interactions between the isolated species in solution phase. The UV-Vis spectra of 1·Cl and 1·Br exhibited principal absorptions at 262 and 300 nm (Fig. S18b, ESI†), whereas in the IR, a complex profile of the phenyl ring and intense absorptions from the CF3 groups in the region 1350–1000 cm−1 were observed. Notably, another distinct sharp absorption at 679 cm−1 consistent with the characteristic tellurane (Te–O) stretch was also seen for both 1·Cl and 1·Br (Fig. S18c and S15, ESI†).37 Interestingly, at wavenumbers larger than 1350 cm−1, 1·Cl and 1·Br demonstrated excellent IR transparency, importantly indicating the absence of hydroxyl functionalities (Fig. 18c, ESI†).38 ESI-MS analysis of 1·Cl and 1·Br revealed the presence of several telluroxane cations including; [Ar2TeO]+ (m/z = 572.9), [(Ar2Te)2O2]+ (m/z = 1140.8), [(Ar2Te)2O2]+ (m/z = 1140.8) and [(Ar2Te)3O3]+ (m/z = 1712.9), which are presumably the result of autoionization under MS conditions (Fig. S16, ESI†).28 Determined melting points of 1·Cl and 1·Br (47 °C and 49 °C respectively) are in stark contrast with the vast majority of reported telluroxane structures, which typically exhibit high melting points (>100 °C).26,28,38,39 The combined spectroscopic evidence, such as, resolved concentration independent NMR signals and physical properties including low sharp melting points and excellent solubility in a wide range of non-polar solvents (e.g. dichloromethane, hexane, toluene, Fig. S4–S6, ESI†) all suggests that both 1·Cl and 1·Br are discrete molecular entities. It is noteworthy that this is contrary to the vast majority of telluroxanes reported to date, which are invariably oligomeric or polymeric structures of largely ionic character.
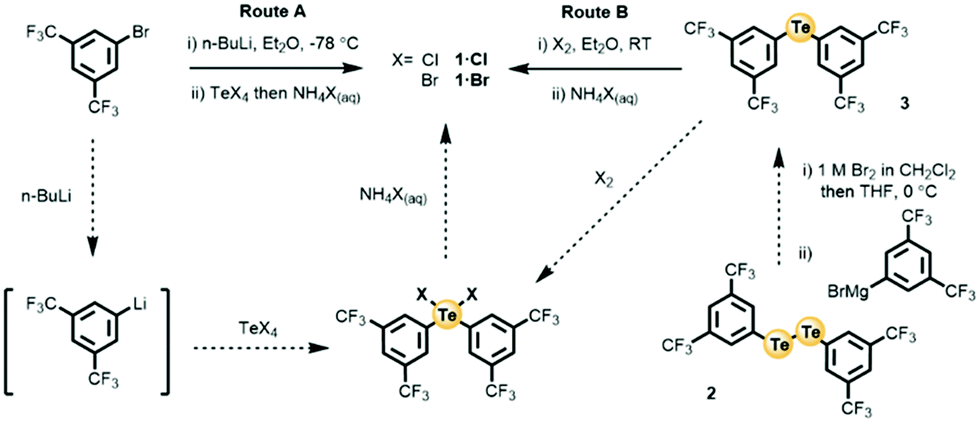 | ||
| Scheme 1 Synthetic routes A and B to organotelluroxanes 1·Cl and 1·Br. | ||
Crystals of 1·Cl and 1·Br suitable for X-ray diffraction analysis were obtained by slow evaporation of chloroform solutions (see ESI† for full discussion of the crystallographic data analysis). Fig. 2 shows both 1·Cl and 1·Br exhibit impressive isostructural oblate-shaped cluster structures, consisting of two orthogonally arranged tetratelluroxane chains. These constituent oligotelluroxane chains are comprised of internal ditellurane (O–TeAr2–O–TeAr2–O) and two terminal telluronium (TeAr2–O) units. The terminal Te atoms of the chain are connected to Te termini of the other oligotelluroxane fragment through two halide bridging ligands, generating a macrotricyclic Te8O6X4 core. This internal oxotellurium halide network is shielded by an array of exterior bis-trifluoromethylphenyl substituents attached to the Te(IV) centres. Inspection of the telluroxane cluster structures aids the rationalisation of several observations made during their synthesis. Firstly, the absence of any product formation without the presence of ammonium chloride or bromide, indicates that the cluster assembly is facilitated by a high concentration of chloride or bromide, which is understandable if the halide serves as a templating agent. Presumably, the inability to form an analogous structure with an iodide counteranion is attributed to the decreased anion basicity and/or structural implications imposed on the framework by the larger halide. The high solubility in organic solvents such as hexane and toluene can also be explained, as the polar inorganic oxotellurium halide interior is effectively shielded by multiple fluorinated aromatic residues. Motivated by these findings we sought to investigate if this organic envelopment of the Te8O6X4 core conferred stability to the cluster assembly. In this vein, samples of 1·Cl and 1·Br were dissolved in CD2Cl2 to which was added a 10-fold excess of a AgPF6 or NaBArF4 in CD2Cl2. Remarkably, the persistent solution homogeneity and negligible difference in the pre- and post- salt addition 1H NMR spectra, even after 30 minutes of stirring at room temperature, indicated the cluster structures possess considerable resistance to anion exchange reactions. This may be interpreted as a combination of kinetic inertness, arising from the inaccessibility of the halide containing core, and a thermodynamic penalty from the rupture of a bifurcated ChB–halide bond. Attention subsequently turned to revisiting 125Te NMR characterisation of the clusters. At 298 K an acetone-d6 solution of 1·Cl and 1·Br exhibits a single clearly resolved signal in the 125Te spectrum, at a chemical shift consistent with a typical tellurane environment. However, the structure as determined by XRD reveals the presence of two tellurium environments, namely the tellurane and telluronium. The absence of a resonance corresponding to the telluronium termini at room temperature conditions is not entirely unexpected and is consistent with numerous reports of oligotelluroxanes.39 Attempts to observe the characteristically broad telluronium signal by recording the 125Te spectra of 1·Cl acetone-d6 at −90 °C did not resolve any additional Te signals (see ESI†), despite high concentrations (50 mM) and long acquisition times (10 hours). Interestingly, the corresponding 1H and 19F NMR spectra at −90 °C, also revealed only minor changes at the low temperature. These low temperature NMR spectroscopic observations suggest that even at −90 °C the rotation of bis-trifluoromethyl aryl substituents is still fast on the 1H and 19F NMR timescales. To further probe the chemical integrity of the clusters, water stability studies were conducted wherein increasing water volumes were added to THF solutions of either 1·Cl or 1·Br (10−5 M). Importantly, no significant perturbations in the UV-Vis spectrum were observed with increasing water fractions, indicating appreciable cage stability towards hydrolysis. However, during the course of these experiments, it was observed that as the water percentage increased the solutions became increasingly emissive. Intrigued by this behaviour, we sought to investigate the photophysical properties of 1·Cl, and how they might be affected by aggregation. Preliminary fluorescence studies demonstrated that a THF solution of 1·Cl (10−5 M) is essentially non-emissive (λex = 350 nm). However, upon increasing the percentage water fraction (fw) of the solution (0–50%), a progressive increase in fluorescence intensity is observed (λmax = 350 nm), reaching a dramatic 13-fold increase in emission intensity at fw = 50% (Fig. S9, ESI†). This type of solvent dependent emission behaviour is characteristic of aggregation induced emissive (AIE) molecules, in which upon aggregation the restriction of intramolecular rotational and or vibrational freedom supresses non-radiative decay pathways, thereby increasing fluorescent output. Analogous experiments conducted with 1·Br also revealed similar AIE like behaviour, suggesting that the identity of the halide plays little or no role in the cluster's photophysical behaviour.
 | ||
| Fig. 2 Solid state structure of (a) 1·Cl and (b) 1·Br. (c) Structure of the oxytellurium halide core Te8O6X4. (d) Chemical structure of Ar16Te8O6X4. (e) Space filling representation of 1·Cl. Grey = carbon, light green = fluorine, green = chloride, brown = bromine, red = oxygen, orange = tellurium. | ||
In conclusion, we report the synthesis of two novel organotelluroxane Ar16Te8O6X4 molecular clusters. It is shown that the directed assembly and structural integrity relies upon the formation of potent Te⋯X− (X− = Cl−, Br−) ChB-anion template interactions. Importantly as demonstrated by physical properties, multinuclear NMR characterisation and reactivity studies, the hydrophobic insulation of the inorganic oxotellurium halide core by an organic exterior confers remarkable stability to the cluster framework. In addition, preliminary fluorescence investigations demonstrate 1·Cl and 1·Br exhibit AIE behaviour in THF–H2O mixtures. In the case of 1·Cl an impressive 13-fold enhancement in emission intensity at a fw value of 50% is observed. These results serve to illustrate the powerful strategy of exploiting potent ChB–anion interactions in the design and construction of elaborate main-group supramolecular inorganic–organic hypervalent Te(IV) assemblies, potentially displaying a wealth of structural diversity and optical/electronic material properties.
A. D thanks the EPSRC for a studentship (Grant reference number EP/N509711/1). A. J. M. M thanks the MICIN and FEDER (RYC-2017-21783, EUR2020-112189, PID2019-108292RA-I00 and P20-00373) and AIQBE (55-2020).
Conflicts of interest
There are no conflicts to declare.Notes and references
- S. Leininger, B. Olenyuk and P. J. Stang, Chem. Rev., 2000, 100, 853–908 CrossRef CAS PubMed.
- J. E. M. Lewis, P. D. Beer, S. J. Loeb and S. M. Goldup, Chem. Soc. Rev., 2017, 46, 2577–2591 RSC.
- M. Fujita, M. Tominaga, A. Hori and B. Therrien, Acc. Chem. Res., 2005, 38, 369–378 CrossRef CAS PubMed.
- M. Han, D. M. Engelhard and G. H. Clever, Chem. Soc. Rev., 2014, 43, 1848–1860 RSC.
- K. K. G. Wong, N. H. Pérez, A. J. P. White and J. E. M. Lewis, Chem. Commun., 2020, 56, 10453–10456 RSC.
- J. F. Nierengarten, C. O. Dietrich-Buchecker and J. P. Sauvage, J. Am. Chem. Soc., 1994, 116, 375–376 CrossRef CAS PubMed.
- L. Zhang, D. P. August, J. Zhong, G. F. S. Whitehead, I. J. Vitorica-Yrezabal and D. A. Leigh, J. Am. Chem. Soc., 2018, 140, 4982–4985 CrossRef CAS PubMed.
- L.-J. Riwar, N. Trapp, K. Root, R. Zenobi and F. Diederich, Angew. Chem., Int. Ed., 2018, 57, 17259–17264 CrossRef CAS PubMed.
- N. Biot and D. Bonifazi, Chem. – Eur. J., 2018, 24, 5439–5443 CrossRef CAS PubMed.
- A. Docker, X. Shang, D. Yuan, H. Kuhn, Z. Zhang, J. J. Davis, P. D. Beer and M. J. Langton, Angew. Chem., Int. Ed., 2021, 60, 19442–19450 CrossRef CAS PubMed.
- K. Kobayashi, H. Izawa, K. Yamaguchi, E. Horn and N. Furukawa, Chem. Commun., 2001, 1428–1429 RSC.
- N. Biot, D. Romito and D. Bonifazi, Cryst. Growth Des., 2021, 21, 536–543 CrossRef CAS PubMed.
- N. Biot and D. Bonifazi, Coord. Chem. Rev., 2020, 413, 213243 CrossRef CAS.
- P. C. Ho, J. Lomax, V. Tomassetti, J. F. Britten and I. Vargas-Baca, Chem. – Eur. J., 2021, 27, 10849–10853 CrossRef CAS PubMed.
- P. C. Ho, J. Z. Wang, F. Meloni and I. Vargas-Baca, Coord. Chem. Rev., 2020, 422, 213464 CrossRef CAS.
- N. A. Puskarevsky, A. I. Smolentsev, A. A. Dmitriev, I. Vargas-Baca, N. P. Gritsan, J. Beckmann and A. V. Zibarev, Chem. Commun., 2020, 56, 1113–1116 RSC.
- J. Lee, L. M. Lee, Z. Arnott, H. Jenkins, J. F. Britten and I. Vargas-Baca, New J. Chem., 2018, 42, 10555–10562 RSC.
- N. A. Pushkarevsky, P. A. Petrov, D. S. Grigoriev, A. I. Smolentsev, L. M. Lee, F. Kleemiss, G. E. Salnikov, S. N. Konchenko, I. Vargas-Baca, S. Grabowsky, J. Beckmann and A. V. Zibarev, Chem. – Eur. J., 2017, 23, 10987–10991 CrossRef CAS PubMed.
- P. C. Ho, H. A. Jenkins, J. F. Britten and I. Vargas-Baca, Faraday Discuss., 2017, 203, 187–199 RSC.
- P. C. Ho, J. Rafique, J. Lee, L. M. Lee, H. A. Jenkins, J. F. Britten, A. L. Braga and I. Vargas-Baca, Dalton Trans., 2017, 46, 6570–6579 RSC.
- P. C. Ho, P. Szydlowski, J. Sinclair, P. J. W. Elder, J. Kübel, C. Gendy, L. M. Lee, H. Jenkins, J. F. Britten, D. R. Morim and I. Vargas-Baca, Nat. Commun., 2016, 7, 11299 CrossRef CAS PubMed.
- P. C. Ho, V. Tomassetti, J. F. Britten and I. Vargas-Baca, Inorg. Chem., 2021, 60, 16726–16733 CrossRef CAS PubMed.
- J. Beckmann and P. Finke, in Selenium Tellurium Chemistry Small Molecules Biomolecules Materials, ed. J. D. Woollins, R. Laitinen, Springer, Berlin, Heidelberg, 2011, pp. 151–177 Search PubMed.
- K. Srivastava, A. Panda, S. Sharma and H. B. Singh, J. Organomet. Chem., 2018, 861, 174–206 CrossRef CAS.
- T. M. Klapötke, B. Krumm, P. Mayer and O. P. Ruscitti, Z. Naturforsch., B: J. Chem. Sci., 2002, 57, 145–150 CrossRef.
- K. Srivastava, S. Sharma, H. B. Singh, U. P. Singh and R. J. Butcher, Chem. Commun., 2010, 46, 1130–1132 RSC.
- H. Citeau, K. Kirschbaum, O. Conrad and D. M. Giolando, Chem. Commun., 2001, 2006–2007 RSC.
- L. Kirsten, J. F. Rodrigues, A. Hagenbach, A. Springer, N. R. Pineda, P. C. Piquini, M. R. Jungfer, E. S. Lang and U. Abram, Angew. Chem., 2021, 133, 15645–15651 CrossRef.
- A. Docker, T. Bunchuay, M. Ahrens, A. J. Martinez-Martinez and P. D. Beer, Chem. – Eur. J., 2021, 27, 7837–7841 CrossRef CAS PubMed.
- Y. C. Tse, A. Docker, Z. Zhang and P. D. Beer, Chem. Commun., 2021, 57, 4950–4953 RSC.
- L. E. Bickerton, A. Docker, A. J. Sterling, H. Kuhn, F. Duarte, P. D. Beer and M. J. Langton, Chem. – Eur. J., 2021, 27, 11738–11745 CrossRef CAS PubMed.
- A. Docker, J. G. Stevens and P. D. Beer, Chem. – Eur. J., 2021, 27, 14600–14604 CrossRef CAS PubMed.
- T. Bunchuay, A. Docker, U. Eiamprasert, P. Surawatanawong, A. Brown and P. D. Beer, Angew. Chem., Int. Ed., 2020, 59, 12007–12012 CrossRef CAS PubMed.
- A. Docker, C. H. Guthrie, H. Kuhn and P. D. Beer, Angew. Chem., Int. Ed., 2021, 60, 21973–21978 CrossRef CAS PubMed.
- B. Zhou and F. P. Gabbaï, Chem. Sci., 2020, 11, 7495–7500 RSC.
- B. Zhou and F. P. Gabbaï, J. Am. Chem. Soc., 2021, 143, 8625–8630 CrossRef CAS PubMed.
- N. W. Alcock and W. D. Harrison, J. Chem. Soc., Dalton Trans., 1982, 1421–1428 RSC.
- R. Deka, A. Sarkar, R. J. Butcher, P. C. Junk, D. R. Turner, G. B. Deacon and H. B. Singh, Dalton Trans., 2020, 49, 1173–1180 RSC.
- J. Beckmann, J. Bolsinger and A. Duthie, Chem. – Eur. J., 2011, 17, 930–940 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2131078. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d2cc00320a |
| This journal is © The Royal Society of Chemistry 2022 |