
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Increasing the oxidation power of TCNQ by coordination of B(C6F5)3†
Paul Anton
Albrecht
 a,
Susanne Margot
Rupf
a,
Malte
Sellin
ab,
Johanna
Schlögl
a,
Sebastian
Riedel
a and
Moritz
Malischewski
*a
a,
Susanne Margot
Rupf
a,
Malte
Sellin
ab,
Johanna
Schlögl
a,
Sebastian
Riedel
a and
Moritz
Malischewski
*a
aFreie Universität Berlin, Institute of Chemistry and Biochemistry, Fabeckstr. 34-36, 14195 Berlin, Germany. E-mail: moritz.malischewski@fu-berlin.de
bAlbert-Ludwigs-Universität Freiburg, Institute of Inorganic and Analytical Chemistry, Albertstraße 21, 79104 Freiburg, Germany
First published on 17th March 2022
Abstract
The oxidation power of the cyanocarbon TCNQ (tetracyano-quinodimethane) can be significantly increased to approximately E = +0.9 V vs. Cp2Fe by coordination of up to four equivalents of the strong fluorinated Lewis acid B(C6F5)3, resulting in a highly reactive but easy-to-use oxidation system. Thianthrene and tris(4-bromophenyl)amine were oxidized to the corresponding radical cations. Dianionic [TCNQ·4 B(C6F5)3]2− was formed upon reduction with two equivalents of ferrocene or decamethylcobaltocene. [TCNQ·4 B(C6F5)3]− and [TCNQ·4 B(C6F5)3]2− are rare cases of redox-active weakly-coordinating anions.
Strong one-electron oxidizing agents are of great synthetic utility, providing access to highly reactive target molecules. However, it is challenging to find a good balance between oxidation power, absence of side reactions, ease of handling and availability of the reagents. For example, inorganic fluorine compounds like (gaseous) AsF5 or metal hexafluorides are extremely powerful oxidants e.g. in liquid SO2, but they can often not be handled in standard laboratories.1,2 In their seminal review on chemical redox agents, Connelly and Geiger discussed more popular reagents, e.g. Ag+ and NO+, ferrocenium and triarylaminium salts.3 They classified all reagents with redox potentials E > +0.8 V vs. Cp2Fe as very strong oxidants. However, it has to be stated, that especially in combination with transition metal complexes, the use of Ag+ or NO+ can lead to side reactions.4,5 Interestingly, the oxidation power of Ag+ can be significantly increased in combination with elemental halogens X2 (X = Cl, Br, I), since the formation of insoluble AgX provides an additional driving force.6,7 Another highly useful system was developed by Poleschner. By using XeF2 in combination with fluoride acceptors (BF3·Et2O, Me3SiOTf, etc.) different weakly-coordinating anions can be introduced.8 Jenne demonstrated the very high oxidation power of the boron cluster radical Me3NB12Cl11, however its multi-step synthesis seems to have prevented widespread use by other groups.9 The same is true for Michl's CB11Me12 or a perfluorinated ammoniumyl cation of the Krossing group.10,11 In contrast to this, cyanocarbons, although mild oxidants, are often commercially available and air-stable. Due to the electron-withdrawing character of the cyano groups, neutral cyanocarbons are molecules with high electron affinities.12 Consequently, they easily form radical anions (or even dianions) of significant stability when treated with a reducing agent. This has led to a plethora of applications, e.g. in molecular magnetism13,14 and for the doping of organic polymers.15 Since tetracyanoquinodimethane (TCNQ) is only a mild oxidant (first reduction potential E = −0.30 V, second reduction potential E = −0.88 V vs. Cp2Fe)3,16,17 we assumed that its electron affinity could be increased and manipulated by coordination of Lewis acids to the terminal nitrogen atoms.18
Tris(pentafluorophenyl)boron is a commercially available and potent Lewis acid.19 It has a high affinity for hard donor atoms, e.g. nitrogen, but is typically non-oxidizing.20,21 Indeed, the reduction to its radical anion is only achieved at redox potentials below E = −1.7 V vs. Cp2Fe.22–24 Interestingly, it has been shown by Stephan and Erker that the combination of two equivalents of B(C6F5)3 with the electron acceptor p-benzoquinone C6H4O2 is able to oxidize decamethylferrocene to its cation.25,26 Depending on the stoichiometry [C6H4O2·2 B(C6F5)3]−/2− mono- or dianions are obtained. By choosing other Lewis acids, the oxidation power of the system can be tuned.27 Another example for cooperative effects between electron acceptors and fluorinated Lewis acids is the combination of dioxygen with two equivalents of B(C6F5)3 which is even able to oxidize air-stable ferrocene to ferrocenium under formation of [(C6F5)3BOOB(C6F5)3]2− dianions.28 In the area of coordination chemistry, seminal works by Fukuzumi have shown the importance of Lewis acidic metal centers in dioxygen activation.29–31
In order to develop a new oxidizing system, we investigated the combination of TCNQ with four equivalents of B(C6F5)3 (Scheme 1).32 Although we were not able to isolate a neutral 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4 adduct (due to the precipitation of the 1:2 adduct), this mixture is strongly oxidizing in weakly-basic solvents like dichloromethane or o-dichlorobenzene. Both tris(4-bromophenyl)amine (E = +0.70 V vs. Cp2Fe) and thianthrene (E = +0.86 V vs. Cp2Fe) are instantaneously oxidized to their corresponding (blue/violet) radical cations,3 forming monoanionic [TCNQ·4 B(C6F5)3]− as the counterion. Both [N(C6H4Br)3]+ [TCNQ·4 B(C6F5)3]− ·4 o-C6H4Cl2 and [(C6H4)2S2]+ [TCNQ·4 B(C6F5)3]− were structurally characterized via single crystal X-ray diffraction (Fig. 1).33 Due to the 4:1 stoichiometry, all cyano groups are coordinated to the Lewis acidic boron centers. Consequently, no potential nitrogen donor sites are present,34 resulting in a new, large weakly-coordinating monoanion with 60 fluorine atoms involved. To estimate the reduction potential that is needed to reduce the radical monoanions to diamagnetic dianions, we successfully used two equivalents of ferrocene per TCNQ. Their oxidation to ferrocenium showed the significant oxidation power of the radical monoanions. Additionally, this indicates potential stability of the dianion against dioxygen. Nevertheless these adducts are sensitive to water and Lewis bases. Despite its low solubility in dichloromethane, the structure of [Cp2Fe]+2 [TCNQ·4 B(C6F5)3]2−·2 CH2Cl2 could be determined via single-crystal X-ray diffraction.33 To prepare a salt with diamagnetic cations and anions (and better solubility), two equivalents of decamethylcobaltocene were successfully used as reducing agent. Correspondingly, the structure of [Cp*2Co]+2 [TCNQ·4 B(C6F5)3]2−·2 CH2Cl2 was also determined.33 In all four characterized salts, the structures of the cations are in accordance with literature reports and will not be further discussed here. H⋯F contacts between cations and anions are in the range of 2.4–2.7 Å and can therefore be considered as weak hydrogen bond contacts. In general, the mono- and dianions look very similar at first sight since the four cyano groups are slightly tilted out of the plane of the central six-membered ring (angles 4.75°–11.21°). However, several characteristic trends regarding specific bond lengths of the TCNQ core can be noticed. Neutral TCNQ displays significant variations regarding the C–C bond lengths in its six-membered ring due to its quinoidal nature. However, upon reduction to its mono- and (aromatic) dianion, these differences are becoming less pronounced.35 The same trend is observed for the adducts [TCNQ·4 B(C6F5)3]0/−/2− (Fig. 2, Table. 1). The six-membered ring in [(C6H4)2S2]+ [TCNQ·4 B(C6F5)3]− is characterized by two short (1.359(3) Å) and four long bonds (1.414(3)–1.426(3) Å. The C–C(CN)2 bond has a length of 1.426(3) Å. In [Cp2Fe]+2 [TCNQ·4 B(C6F5)3]2−·2 CH2Cl2 the C–C bonds of the six-membered ring of TCNQ are all very similar in length (1.383(2)–1.396(2) Å), indicating a more effective electron delocalization. The corresponding C–C bond to the C(CN)2 group is longer (1.480(2) Å) than in the monoanion. In the ferrocenium salt the average C
:4 adduct (due to the precipitation of the 1:2 adduct), this mixture is strongly oxidizing in weakly-basic solvents like dichloromethane or o-dichlorobenzene. Both tris(4-bromophenyl)amine (E = +0.70 V vs. Cp2Fe) and thianthrene (E = +0.86 V vs. Cp2Fe) are instantaneously oxidized to their corresponding (blue/violet) radical cations,3 forming monoanionic [TCNQ·4 B(C6F5)3]− as the counterion. Both [N(C6H4Br)3]+ [TCNQ·4 B(C6F5)3]− ·4 o-C6H4Cl2 and [(C6H4)2S2]+ [TCNQ·4 B(C6F5)3]− were structurally characterized via single crystal X-ray diffraction (Fig. 1).33 Due to the 4:1 stoichiometry, all cyano groups are coordinated to the Lewis acidic boron centers. Consequently, no potential nitrogen donor sites are present,34 resulting in a new, large weakly-coordinating monoanion with 60 fluorine atoms involved. To estimate the reduction potential that is needed to reduce the radical monoanions to diamagnetic dianions, we successfully used two equivalents of ferrocene per TCNQ. Their oxidation to ferrocenium showed the significant oxidation power of the radical monoanions. Additionally, this indicates potential stability of the dianion against dioxygen. Nevertheless these adducts are sensitive to water and Lewis bases. Despite its low solubility in dichloromethane, the structure of [Cp2Fe]+2 [TCNQ·4 B(C6F5)3]2−·2 CH2Cl2 could be determined via single-crystal X-ray diffraction.33 To prepare a salt with diamagnetic cations and anions (and better solubility), two equivalents of decamethylcobaltocene were successfully used as reducing agent. Correspondingly, the structure of [Cp*2Co]+2 [TCNQ·4 B(C6F5)3]2−·2 CH2Cl2 was also determined.33 In all four characterized salts, the structures of the cations are in accordance with literature reports and will not be further discussed here. H⋯F contacts between cations and anions are in the range of 2.4–2.7 Å and can therefore be considered as weak hydrogen bond contacts. In general, the mono- and dianions look very similar at first sight since the four cyano groups are slightly tilted out of the plane of the central six-membered ring (angles 4.75°–11.21°). However, several characteristic trends regarding specific bond lengths of the TCNQ core can be noticed. Neutral TCNQ displays significant variations regarding the C–C bond lengths in its six-membered ring due to its quinoidal nature. However, upon reduction to its mono- and (aromatic) dianion, these differences are becoming less pronounced.35 The same trend is observed for the adducts [TCNQ·4 B(C6F5)3]0/−/2− (Fig. 2, Table. 1). The six-membered ring in [(C6H4)2S2]+ [TCNQ·4 B(C6F5)3]− is characterized by two short (1.359(3) Å) and four long bonds (1.414(3)–1.426(3) Å. The C–C(CN)2 bond has a length of 1.426(3) Å. In [Cp2Fe]+2 [TCNQ·4 B(C6F5)3]2−·2 CH2Cl2 the C–C bonds of the six-membered ring of TCNQ are all very similar in length (1.383(2)–1.396(2) Å), indicating a more effective electron delocalization. The corresponding C–C bond to the C(CN)2 group is longer (1.480(2) Å) than in the monoanion. In the ferrocenium salt the average C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N bond length (1.149(2) Å) is very similar to the corresponding value of the thianthrenium salt (1.145(3) Å). The changes of the B–N bond lengths are more significant: 1.550(2)–1.561(2) Å in the dianions compared to 1.580(3)–1.590(3) Å in the monoanions, indicating a stronger donor–acceptor interaction in the dianions.
N bond length (1.149(2) Å) is very similar to the corresponding value of the thianthrenium salt (1.145(3) Å). The changes of the B–N bond lengths are more significant: 1.550(2)–1.561(2) Å in the dianions compared to 1.580(3)–1.590(3) Å in the monoanions, indicating a stronger donor–acceptor interaction in the dianions.
 | ||
| Scheme 1 Substrate scope of the new oxidation system TCNQ + 4 B(C6F5)3. | ||
 | ||
| Fig. 1 Molecular structure of the radical anion in [(C6H4)2S2]+ [TCNQ·4 B(C6F5)3]−, thermal ellipsoids shown with 50% probability, colour code: hydrogen white, carbon grey, fluorine green, nitrogen blue, boron rosé. | ||
 | ||
| Fig. 2 Lewis formulas of [TCNQ·4 B(C6F5)3] (left) and [TCNQ·4 B(C6F5)3]2− (right) and assignment of the labels for the discussion of bond lengths. | ||
| Bond | [TCNQ·4 B(C6F5)3] calculated | [TCNQ·4 B(C6F5)3]− calculated | [TCNQ·4 B(C6F5)3]− experimental | [TCNQ·4 B(C6F5)3]2− calculated | [TCNQ·4 B(C6F5)3]2− experimental |
|---|---|---|---|---|---|
| a | 1.358–1.365 | 1.371–1.378 | 1.359(3)–1.364(6) | 1.391–1.394 | 1.383(2)–1.388(2) |
| b | 1.440–1.444 | 1.425–1.426 | 1.411(6)–1.426(6) | 1.406–1.407 | 1.396(2)–1.399(2) |
| c | 1.401–1.402 | 1.431–1.433 | 1.426(6)–1.431(3) | 1.477–1.480 | 1.470(2)–1.480(2) |
| d | 1.412–1.416 | 1.401–1.405 | 1.397(6)–1.405(6) | 1.383–1.386 | 1.382(2)–1.385(2) |
| e | 1.151–1.154 | 1.154–1.156 | 1.140(6)–1.145(3) | 1.164–1.166 | 1.147(2)–1.151(2) |
| f | 1.574–1.582 | 1.562–1.588 | 1.580(6)–1.589(3) | 1.550–1.561 | 1.550(2)–1.561(2) |
Quantum-chemical calculations at the DFT level B3LYP-D3(BJ)/Def2-SVP have been performed to evaluate the structure and the vibrational frequencies of the neutral, mono- and dianionic 4:1 adducts between B(C6F5)3 and TCNQ. They confirm that, upon reduction, the C–C bond lengths of the six-membered ring become less variable. Additionally, a slight increase of CN bond lengths is predicted upon reduction of the neutral species to the mono- and dianion (1.152 Å → 1.156 Å → 1.165 Å). Principally, one would expect a decrease of the computed ![[small nu, Greek, tilde]](https://www.rsc.org/images/entities/i_char_e0e1.gif) (CN) frequency for the reduced species. In general, four different ν(CN) vibrations are predicted within the range of 2220–2350 cm−1. Upon reduction, the red-shift of the symmetric (CN) vibration (which is highest in energy) is relatively little pronounced in the series (2348 → 2345 → 2317 cm−1). In contrast, the shift of the antisymmetric (CN) vibration (which is lowest in energy) is much more pronounced (2311 → 2290 → 2221 cm−1). Indeed, the experimental IR spectra of salts that contain the monoanions have two bands at 2290 and ≈2250 cm−1, while two bands at 2284 and ≈ 2205 cm−1 are observed for the dianion (see Fig. S9, ESI†). The widening of the range of (CN) frequencies upon reduction is consequently both experimentally and computationally found. Attempts to measure Raman spectra of the products were unsuccessful due to intense fluorescence or decomposition by laser light.
(CN) frequency for the reduced species. In general, four different ν(CN) vibrations are predicted within the range of 2220–2350 cm−1. Upon reduction, the red-shift of the symmetric (CN) vibration (which is highest in energy) is relatively little pronounced in the series (2348 → 2345 → 2317 cm−1). In contrast, the shift of the antisymmetric (CN) vibration (which is lowest in energy) is much more pronounced (2311 → 2290 → 2221 cm−1). Indeed, the experimental IR spectra of salts that contain the monoanions have two bands at 2290 and ≈2250 cm−1, while two bands at 2284 and ≈ 2205 cm−1 are observed for the dianion (see Fig. S9, ESI†). The widening of the range of (CN) frequencies upon reduction is consequently both experimentally and computationally found. Attempts to measure Raman spectra of the products were unsuccessful due to intense fluorescence or decomposition by laser light.
Based on DFT calculations (B3LYP-D3(BJ)/Def2-SVP) the adiabatic electron affinities of the neutral adduct was estimated to be 583 kJ mol−1 (6.04 eV). The reduction of the corresponding monoanion to the dianion is also highly favourable (310 kJ mol−1, 3.21 eV). In comparison, free TCNQ has an experimental electron affinity of 3.38 eV.36 A similar trend can be seen when LUMO energies are taken into account. While the calculated LUMO energy of TCNQ is −4.98 eV, the corresponding value for neutral [TCNQ·4 B(C6F5)3] is −6.80 eV (extension on some C6F5 rings). Consequently, the coordination of Lewis acids clearly increases its oxidation power and underlines our cooperativity concept for such systems.
Cyclic voltammetry of [Cp*2Co]+2 [TCNQ·4 B(C6F5)3]2− in CH2Cl2 allowed the determination of the electrochemical properties of the oxidation system (Fig. 3). At a scan rate of 100 mV s−1, a (pseudo)reversible oxidation at E1/2 = +0.065 V vs. Cp2Fe (for the dianion/monoanion redox couple) and an irreversible process at Epa = +1.226 V vs. Cp2Fe (for the irreversible oxidation to the neutral species) were determined. Large peak separations may be caused by slow electron exchange on the electrode. Additionally, [Cp*2Co]+2 [TCNQ·4 B(C6F5)3]2− had sufficient solubility in CD2Cl2 to allow its characterization via multinuclear NMR spectroscopy. Two signals in the 1H-NMR can be assigned to the TCNQ protons (δ = 6.82 ppm) as well as the methyl protons of decamethylcobaltocenium (δ = 1.65 ppm). In the 11B NMR spectrum a broad signal at δ = −11.5 ppm is assigned to the tetracoordinate boron atoms in [TCNQ·4 B(C6F5)3]2− which is also in agreement with other nitrogen-containing adducts.37 For example, [CH3CN·B(C6F5)3] has a 11B NMR shift of −10.3 ppm (in C6D6).21 In comparison the 11B NMR shift of uncoordinated B(C6F5)3 is observed at +59.2 ppm.38 In the 19F NMR three main signals are observed for the meta, para and ortho fluorine atoms in the C6F5 rings δ = −165.4, −159.3 and −134.8 ppm). These signals are slightly shifted in comparison to uncoordinated B(C6F5)3 (δ = −161.3, −144.2 and −128.4 ppm). Additionally, it was possible to find and assign almost all signals in the 13C NMR spectrum. The decamethylcobaltocenium cation gives two signals at δ = 8.2 and 94.6 ppm which is in agreement with literature values.39 For the central TCNQ moiety in [TCNQ·4 B(C6F5)3]2− as well as for the pentafluorophenyl rings four signals are each expected. However, only seven peaks could be detected. The proton-decoupled 13C NMR spectrum displays three doublets for the fluorine-bound carbon atoms of the pentafluorophenyl rings (δ = 148.6; 140.4 and 137.6 ppm). Additionally, one broad signal at δ = 118.3 ppm was observed. These signals are slightly shifted in comparison to free B(C6F5)3 (δ = 149.1 (ortho), 145.8 (para), 138.3 (meta) and 113.8 (ipso) ppm).38 Two additional signals at δ = 126.0 and 123.1 ppm belong clearly to the aromatic TCNQ core of the dianion. In the uncomplexed neutral TCNQ the chemical shifts differ more significantly: δ = 132.6 and 151.7 ppm. For uncomplexed TCNQ the chemical shift of the cyano group is δ = 112.6 ppm.40 However, no corresponding signal was found. Interestingly, one signal of the [TCNQ·4 B(C6F5)3]2− dianion is strongly high-field shifted: the carbon atom of the TCNQ moiety that is bound to the two cyano groups. Due to some carbanion character, the signal is located at δ = 30.5 ppm. In comparison, the same carbon resonates at δ = 90.8 ppm in free TCNQ.
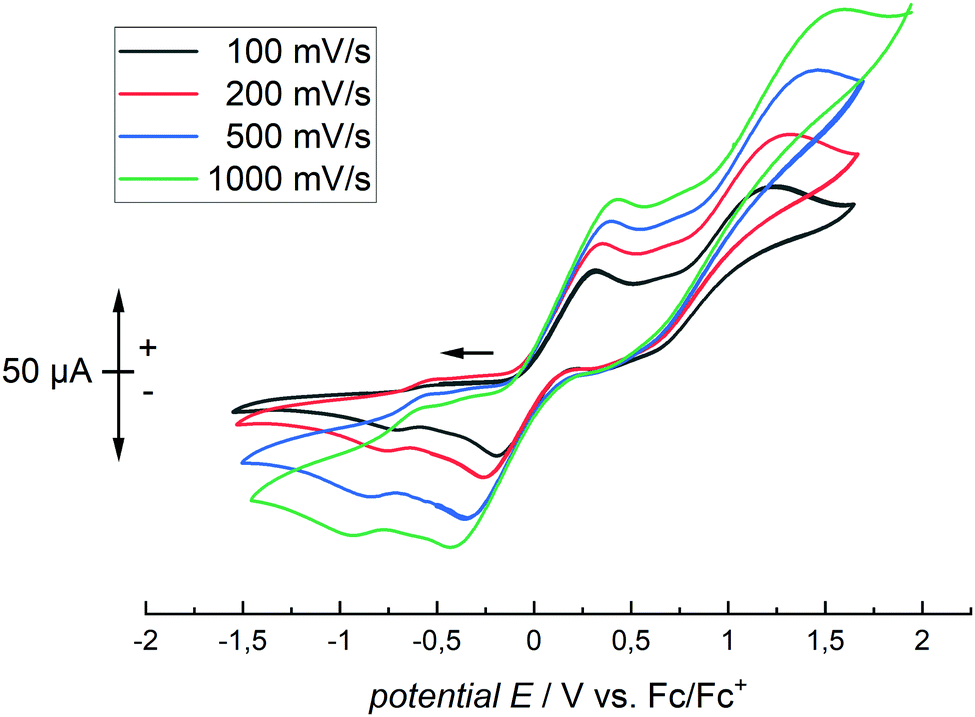 | ||
| Fig. 3 Cyclic voltammogram of [Cp*2Co]+2 [TCNQ·4 B(C6F5)3]2− in CH2Cl2. | ||
EPR spectroscopy at room temperature in dichloromethane was used to prove that electron transfer between the diamagnetic starting materials has taken place. Three experiments were performed: TCNQ with 4 equivalents of B(C6F5)3 was reacted with either tris(4-bromophenylamine), thianthrene or a small amount of ferrocene to generate [Cp2Fe]+ [TCNQ·4 B(C6F5)3]−. In the first two cases EPR spectra showed the presence of two radical species each: the corresponding, well-known radical cations as well as the desired radical anion. In the last case only the spectrum of [TCNQ·4 B(C6F5)3]− was observed, since ferrocenium is not EPR-active at room temperature. The EPR spectrum of the radical anion [TCNQ·4 B(C6F5)3]− consists of one broad, isotropic signal at giso ≈ 2.002 which confirms the strong delocalization of the unpaired electron. Hyperfine splitting was not observed due the large number of spin-carrying nuclei as well as the presence of isotopomers (natural abundance of 10B = 20%, I = 3, and 11B = 80%, I = 3/2).
In summary, we report a potent oxidation system from commercially available chemicals that works in non-coordinating organic solvents and is easy to use. The combination of TCNQ with four equivalents of B(C6F5)3 is able to quantitatively oxidize substrates up to an oxidation potential of E ≈ +0.9 V and generates large, previously unknown weakly-coordinating anions that are redox-active. The resulting [TCNQ·4 B(C6F5)3]− monoanion has still enough oxidation power to oxidize ferrocene to ferrocenium. We are optimistic that similar systems (using cyanocarbons in combination with Lewis acids) could not only be used in molecular chemistry, but will also be of interest for the doping of organic polymers.
Gefördert durch die Deutsche Forschungsgemeinschaft (DFG) – Projektnummer 387284271 – SFB 1349. Computing time was made available by HPC Service of ZEDAT, FU Berlin. We would like to acknowledge the assistance of the Core Facility BioSupraMol supported by the DFG.
Conflicts of interest
There are no conflicts to declare.Notes and references
- M. Malischewski, M. Adelhardt, J. Sutter, K. Meyer and K. Seppelt, Science, 2016, 353, 678–682 CrossRef CAS PubMed.
- R. T. Boeré, S. Kacprzak, M. Keßler, C. Knapp, R. Riebau, S. Riedel, T. L. Roemmele, M. Rühle, H. Scherer and S. Weber, Angew. Chem., Int. Ed., 2011, 50, 549–552 CrossRef PubMed.
- N. G. Connelly and W. E. Geiger, Chem. Rev., 1996, 96, 877–910 CrossRef CAS PubMed.
- M. Schmitt, M. Mayländer, J. Goost, S. Richert and I. Krossing, Angew. Chem., Int. Ed., 2021, 60, 14800–14805 CrossRef CAS PubMed.
- P. J. Malinowski and I. Krossing, Angew. Chem., Int. Ed., 2014, 53, 13460–13462 CrossRef CAS PubMed.
- P. J. Malinowski, D. Himmel and I. Krossing, Angew. Chem., Int. Ed., 2016, 55, 9259–9261 CrossRef CAS PubMed.
- P. J. Malinowski, D. Himmel and I. Krossing, Angew. Chem., Int. Ed., 2016, 55, 9262–9266 CrossRef CAS PubMed.
- H. Poleschner and K. Seppelt, Angew. Chem., Int. Ed., 2013, 52, 12838–12842 CrossRef CAS PubMed.
- P. Bertocco, C. Bolli, J. Derendorf, C. Jenne, A. Klein and K. Stirnat, Chem. – Eur. J., 2016, 22, 16032–16036 CrossRef CAS PubMed.
- B. T. King, B. C. Noll, A. J. McKinley and J. Michl, J. Am. Chem. Soc., 1996, 118, 10902–10903 CrossRef CAS.
- M. Schorpp, T. Heizmann, M. Schmucker, S. Rein, S. Weber and I. Krossing, Angew. Chem., Int. Ed., 2020, 59, 9453–9459 CrossRef CAS PubMed.
- A. L. Farragher and F. M. Page, Trans. Faraday Soc., 1967, 63, 2369–2378 RSC.
- J. S. Miller, A. J. Epstein and W. M. Reiff, Science, 1988, 240, 40–47 CrossRef CAS PubMed.
- J. S. Miller, Chem. Soc. Rev., 2011, 40, 3266–3296 RSC.
- D. Jérome, Chem. Rev., 2004, 104, 5565–5592 CrossRef PubMed.
- D. S. Acker, R. J. Harder, W. R. Hertler, W. Mahler, L. R. Melby, R. E. Benson and W. E. Mochel, J. Am. Chem. Soc., 1960, 82, 6408–6409 CrossRef CAS.
- L. R. Melby, R. J. Harder, W. R. Hertler, W. Mahler, R. E. Benson and W. E. Mochel, J. Am. Chem. Soc., 1962, 84, 3374–3387 CrossRef CAS.
- B. J. McNicholas, R. H. Grubbs, J. R. Winkler, H. B. Gray and E. Despagnet-Ayoub, Chem. Sci., 2019, 10, 3623–3626 RSC.
- G. Erker, Dalton Trans., 2005, 1883–1890 RSC.
- F. Focante, P. Mercandelli, A. Sironi and L. Resconi, Coord. Chem. Rev., 2006, 250, 170–188 CrossRef CAS.
- H. Jacobsen, H. Berke, S. Döring, G. Kehr, G. Erker, R. Fröhlich and O. Meyer, Organometallics, 1999, 18, 1724–1735 CrossRef CAS.
- R. J. Kwaan, C. J. Harlan and J. R. Norton, Organometallics, 2001, 20, 3818–3820 CrossRef CAS.
- S. A. Cummings, M. Iimura, C. J. Harlan, R. J. Kwaan, I. V. Trieu, J. R. Norton, B. M. Bridgewater, F. Jäkle, A. Sundararaman and M. Tilset, Organometallics, 2006, 25, 1565–1568 CrossRef CAS.
- E. J. Lawrence, V. S. Oganesyan, G. G. Wildgoose and A. E. Ashley, Dalton Trans., 2013, 42, 782–789 RSC.
- L. L. Liu, L. L. Cao, Y. Shao and D. W. Stephan, J. Am. Chem. Soc., 2017, 139, 10062–10071 CrossRef CAS PubMed.
- X. Tao, C. G. Daniliuc, R. Knitsch, M. R. Hansen, H. Eckert, M. Lübbesmeyer, A. Studer, G. Kehr and G. Erker, Chem. Sci., 2018, 9, 8011–8018 RSC.
- B. L. Thompson and Z. M. Heiden, Phys. Chem. Chem. Phys., 2021, 23, 9822–9831 RSC.
- J. T. Henthorn and T. Agapie, Angew. Chem., Int. Ed., 2014, 53, 12893–12896 CrossRef CAS PubMed.
- S. Fukuzumi, K. Ohkubo, Y.-M. Lee and W. Nam, Chem. – Eur. J., 2015, 21, 17548–17559 CrossRef CAS PubMed.
- M. Sankaralingam, Y.-M. Lee, W. Nam and S. Fukuzumi, Coord. Chem. Rev., 2018, 365, 41–59 CrossRef CAS.
- T. Devi, Y.-M. Lee, W. Nam and S. Fukuzumi, Coord. Chem. Rev., 2020, 410, 213219 CrossRef CAS.
- P. A. Albrecht, S. M. Rupf, M. Sellin, J. Schlögl, S. Riedel and M. Malischewski, Chem. Rxiv., 2022 DOI:10.26434/chemrxiv-2022-pjm8s.
- P. A. Albrecht, S. M. Rupf, M. Sellin, J. Schlögl, S. Riedel and M. Malischewski, CSD Commun., 2022 Search PubMed (CCDC 2133128, 2133132, 2133133, 2133134).
- R. Choukroun, C. Lorber, D. de Caro and L. Vendier, Organometallics, 2006, 25, 4243–4246 CrossRef CAS.
- T. A. Hudson and R. Robson, Cryst. Growth Des., 2009, 9, 1658–1662 CrossRef CAS.
- G.-Z. Zhu and L.-S. Wang, J. Chem. Phys., 2015, 143, 221102 CrossRef PubMed.
- I. C. Vei, S. I. Pascu, M. L. H. Green, J. C. Green, R. E. Schilling, G. D. W. Anderson and L. H. Rees, Dalton Trans., 2003, 2550–2557 RSC.
- M. Lehmann, A. Schulz and A. Villinger, Angew. Chem., Int. Ed., 2009, 48, 7444–7447 CrossRef CAS PubMed.
- H. Heise, F. H. Köhler, M. Herker and W. Hiller, J. Am. Chem. Soc., 2002, 124, 10823–10832 CrossRef CAS PubMed.
- T. Nunes, A. Vainrub, M. Ribet, F. Rachdi, P. Bernier and M. Almeida, J. Chem. Phys., 1992, 96, 8021–8025 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, spectroscopical and computational data. See DOI: 10.1039/d2cc00314g |
| This journal is © The Royal Society of Chemistry 2022 |