
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTetracyclic silaheterocycle formed through a pericyclic reaction cascade including a two-fold intramolecular C–C bond activation†
Joschua
Helmer
a,
Olli J.
Pakkanen
b,
Chris
Gendy
 b,
Alexander
Hepp
a,
Heikki M.
Tuononen
*b and
Felicitas
Lips
*a
b,
Alexander
Hepp
a,
Heikki M.
Tuononen
*b and
Felicitas
Lips
*a
aInstitut für Anorganische und Analtische Chemie, Corrensstraße 20-30, 48149 Münster, Germany. E-mail: lips@uni-muenster.de
bDepartment of Chemistry, NanoScience Centre, P.O. Box 35, FI-40014 University of Jyväskylä, Finland. E-mail: heikki.m.tuononen@jyu.fi
First published on 7th February 2022
Abstract
Reductive debromination of the tribromoamidosilane 2 gave the tetracyclic silaheterocycle 3 through a unique reaction cascade involving unprecedented two-fold intramolecular cycloaddition by transient silylenes. Experimental and computational analyses of the reaction mechanism allowed the identification of the key intermediates that lead to the silaheterocycle 3 or, alternatively, to the cyclotrisilene 19.
During the past decade, the synthesis and subsequent use of very bulky amido ligands, such as {N(SiR3)Ar} (R = Me, iPr; Ar = aryl group), have paved the way for kinetic stabilisation of many low-valent complexes of heavier main group elements and those in the group 14 particularly.1 This has led to the characterisation of, inter alia, the first examples of amido-substituted digermynes and distannynes with long Ge–Ge or Sn–Sn single bonds, formally bis(tetrylenes),2 stable two-coordinate acyclic silylenes and silylsilylenes,3 and multiply bonded amidodigermynes.4 While these species are of fundamental interest, they also show fascinating reactivity with small molecules. For example, singly bonded amido-substituted digermynes and distannynes activate H2,2 the former even in the solid-state, while acyclic amidosilylenes have been reported to reduce both CO and CO2.5
Recently, we examined the reduction of {N(SiMe3)Dipp}SiBr3 (Dipp = 2,6-iPr2-C6H3) with 1.5 equiv. of activated magnesium (Mg*).6 This was found to give the cluster Si4{N(SiMe3)Dipp}4I, formally a dimer of two amidodisilynes, with a butterfly-type structure.7 Further studies showed that I reacts readily with heavier chalcogens to give amido-substituted cage compounds, while its thermolysis led to the six-vertex silicon cluster Si6{N(SiMe3)Dipp}4II with lone pair character at the ligand-free vertices.8 These results led us to pose the question of the influence of the steric bulk on the structure of I. Specifically, could the butterfly-type Si4 skeleton of I be forced into planarity,9 or would steric strain lead to stabilisation of other amido-substituted silicon rings or cages. As an attempt to answer the above question, we synthesised the tribromoamidosilane {N(SiMe2Ph)Dipp}SiBr32 and carried out its reduction with 1.5 equiv. of Mg*.
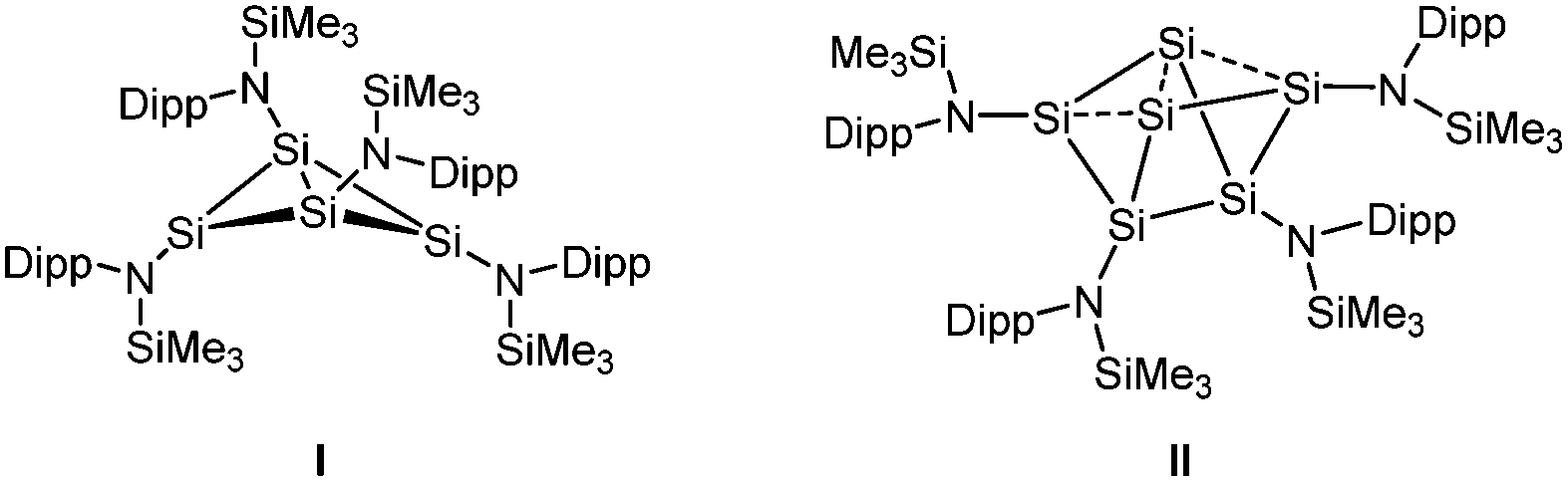
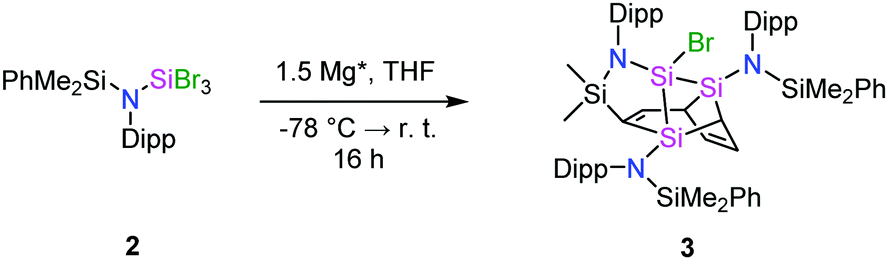
Reaction of the lithium amide Li{N(SiMe2Ph)Dipp} 1 with SiBr4 in Et2O gave the tribromoamidosilane {N(SiMe2Ph)Dipp}SiBr32 in good yield (ESI†), similarly to the corresponding trichlorosilane.10 The NMR data of 2 are consistent with its formulation and its structure was confirmed by single crystal X-ray diffraction (ESI†).
 | ||
| Scheme 1 Reductive debromination of the tribromoamidosilane 2 to the silaheterocycle 3 (Dipp = 2,6-iPr2C6H3). | ||
Compound 2 was reacted with 1.5 equiv. of Mg* in THF (Scheme 1 and ESI†). Extraction with n-hexane gave a yellow residue that contained two primary products based on the NMR data (ESI†). The tetracyclic silaheterocycle 3 was subsequently crystallized as yellow rods from toluene in low but reproducible yields (Fig. 1). The combination of six silicon atoms, three amido groups, and one bromine atom in 3 suggests that the reductive debromination of 2 gives, in sequence, the tetrabromodisilane 4, the dibromodisilene 5, and the disilyne 6 (Scheme 2). This is supported by literature syntheses of [{N(SiMe3)Dipp}SiBr2]2,3a analogous to 4, and [{N(SiMe3)Ar*}Ge]2, similar to 6 but with bulkier substituents (Ar* = C6H2iPr{C(H)Ph2}2-4,2,6, C6H2Me{C(H)Ph2}2-4,2,6).2a,4 Presumably, the conversion of 6 to 3 takes place by a reaction of the former with the bromosilylene 7, generated by an equilibrium with 5, followed by an unprecedented intramolecular two-fold cycloaddition to a single phenyl substituent on one of the SiMe2Ph groups. Dynamic disilene↔silylene equilibria are well-known in the literature.11
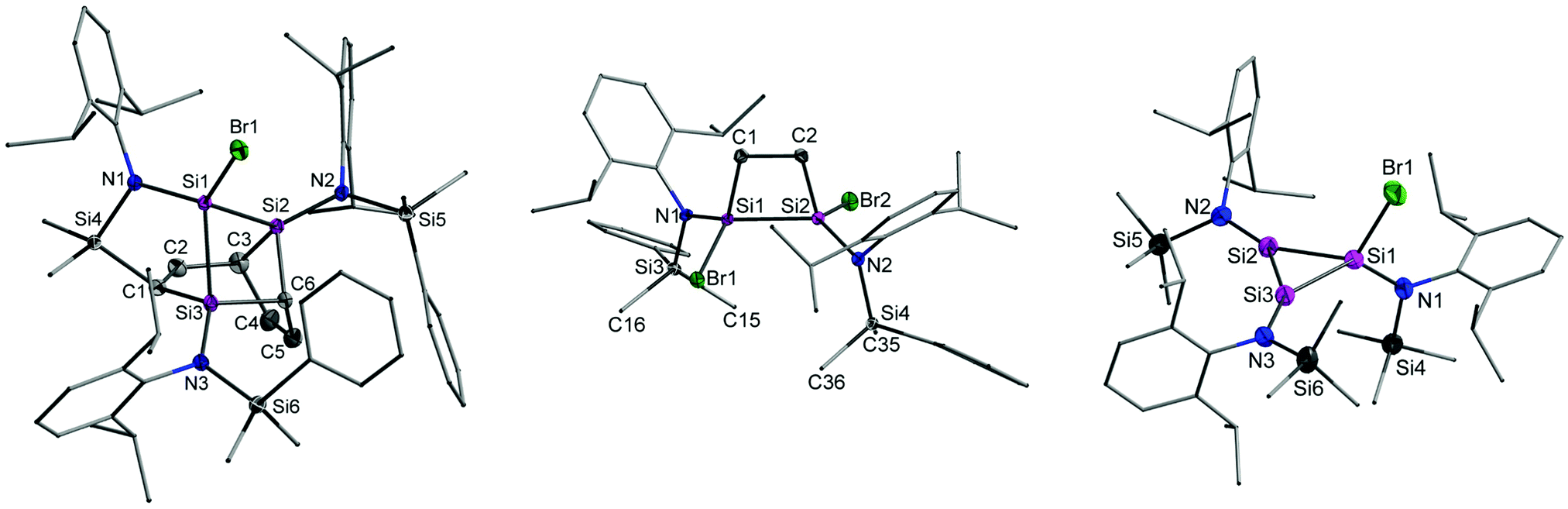 | ||
| Fig. 1 Solid-state structures of 3 (left), 8 (middle), and 19′ (right) with thermal ellipsoids drawn at 50% probability level and hydrogen atoms and co-crystallized solvent molecules omitted for clarity. Full structural details are provided in the ESI.† | ||
 | ||
| Scheme 2 Trapping of intermediates 5 and 7en route from 2 to 3 (Dipp = 2,6-iPr2C6H3, dmbd = 2,3-dimethyl-1,3-butadiene). | ||
Direct experimental support for the steps outlined above was sought by trapping the proposed intermediates. The dibromodisilene intermediate 5 could be trapped through [2+2] cycloaddition with ethylene (Scheme 2 and ESI†), yielding the disilacyclobutane 8 (Fig. 1). A related cycloaddition product has been obtained from reductive dehalogenation of {N(SiMe3)Dipp}SiBr3 with lithium naphthalenide.3a In the presence of 2,3-dimethyl-1,3-butadiene (dmbd), the reductive debromination of 2 gave the [4+2] cycloaddition product {N(SiMe2Ph)Dipp}SiBr(dmbd) 9 as evidence for the equilibrium between 5 and 7 (Scheme 2 and ESI†). Conceivably, 9 could also arise from successive reduction of 2 in the presence of dmbd. Evaluation of the equilibrium between 5 and 7 with computational methods showed that the dibromodisilene is favoured only by 19 kJ mol−1 in the gas phase, supporting the co-existence of 5 and 7 in solution.
Attempts to trap other intermediates, such as 6, were met with failure. For this reason, the reaction mechanism connecting 6 to 3 was investigated computationally. Two related pathways were identified (Scheme 3). In pathway 1, the combination of 6 and 7 gives the acyclic intermediate 10 that then undergoes two consecutive intramolecular cycloadditions coupled by a 1,2-bromine shift to give 3. In pathway 2, an internal cycloaddition of 6 first gives the silacycloheptatriene (silepin) 18 that then reacts with 7 to give 12, an intermediate common to both pathways, that ultimately transforms to 3via 1,2-bromine shift and second cycloaddition.
 | ||
| Scheme 3 Two pathways connecting the disilyne intermediate 6 to the experimentally characterized reaction product 3 (Dipp = 2,6-iPr2C6H3). Calculated relative Gibbs free energies (ΔG and ΔGTS) and Gibbs free energies of activation (ΔG‡) of each individual reaction step are given in kJ mol−1. | ||
The results of DFT calculations (Scheme 3 and ESI†) show that pathways 1 and 2 have low and equal activation barriers (highest ΔG‡ = 59 and 54 kJ mol−1, respectively). However, considering that the initial reduction of 2 is carried out at low temperature, the barrierless and exergonic formation of 10 can give an advantage to pathway 1. The facile nature of this reaction step agrees with the electronic structure of 6 that, like its heavier germanium analogues,2a,4 was found to have two minima in agreement with disilyne and bis(silylene) formulations, that is, with short and long Si–Si bonds, respectively (6short and 6long, ESI†). As the two isomers are separated only by 27 kJ mol−1, with the bis(silylene) structure lower in energy, the reaction between 6 and 7 can be viewed as a combination of two silylenes to generate a disilene functionality. Overall, the computational work indicates that the formation of 3 from 6 should be rapid and exergonic (ΔG = −219 kJ mol−1). The first steps from 6 to 14 are governed almost exclusively by silylene reactivity of the intermediates, whereas the last steps connecting 14 to 3 involve rearrangement of the sila(cyclopropyl) product from the second cycloaddition.
Further support to the pathways outlined in Scheme 3 can be found from the literature. Silylenes are well-known to undergo thermally or photochemically activated intermolecular cycloadditions with several aromatic substrates. These typically give either sila(cyclopropyls) or silepins, such as III and IV, analogous to 11/17 and 12/18.12 Similar reactivity has recently been described for an aluminyl anion stabilised by a xanthene-based diamido ligand that reversibly activates benzene even at room temperature.13 Intermolecular two-fold cycloadditions of silylenes to aromatic frameworks also generate sila(cyclopropyls) and silepins (e.g.III),12a a notable exception being the reaction between a silylene and pyrazine that leads to ring expansion to a cyclooctatriene analogue.14
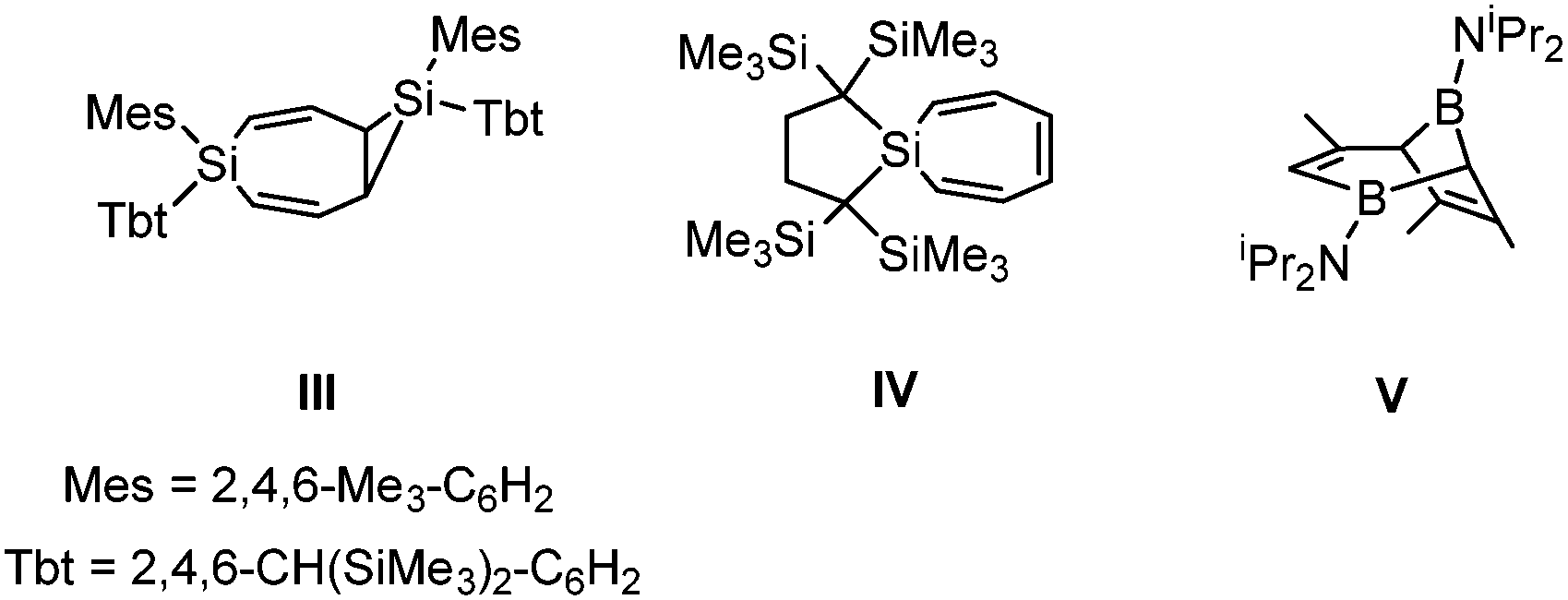
The fact that the conversion of 6 to 3 takes place so readily can be explained by the intramolecular nature of the two-fold cycloaddition and the associated negligible entropic penalty. To our knowledge, there are only two prior examples of intramolecular cycloadditions between silylenes and aromatic Dipp substituents and one example of reversible cycloaddition between a disilene and Cp* (Cp* = C5Me5),15 but 3 is the only example in which the ligand framework is attacked twice. In this respect, the unprecedented structure of 3 is reminiscent of two-fold borylated products, such as V, that have been obtained from defluorination of difluoro(diorganylamino)boranes with Na/K alloy in the presence of benzene derivatives.16 While there are no detailed mechanistic data available, the formation of V is thought to proceed via double attack of in situ generated borylenes on the aromatic substrates, in similar fashion as outlined for the formation of 3 in Scheme 3.
The reaction cascade in Scheme 3 is consistent with our inability to trap 6 or other intermediates en route to 3 using bases, hydrocarbons, or transition metal complexes. In this respect, we also considered the possibility that 7 would undergo an internal cycloaddition analogous to that connecting 6 and 18. Calculations showed, however, that the product is not a stable species on the potential energy surface due to its strained geometry. The same is also true if the cycloaddition would involve a Dipp substituent in place of SiMe2Ph. In similar fashion, detailed potential energy surface scans indicated that the intermediates 17 and 18 will not undergo cycloadditions involving the free silylene moiety and the dangling aromatic substituents adjacent to it. Such reactivity is, however, possible for 13, but in this case, selective attack of the silylene to the silepin is ensured by the negligible activation barrier between 13 and 14 (ΔG‡ = 12 kJ mol−1) and the stability of the product (ΔG = −165 kJ mol−1).
Having examined the most likely pathways available for the intermediates in Scheme 3, we considered the possibility that 10 cyclises to the cyclotrisilene 19. This parallels the reactivity reported for a related base-stabilized disilenyl silylene VI that is in an equilibrium with the corresponding cyclotrisilene VII (and the free base) in solution.17 The results of computational work showed that the ease of cyclisation of 10 depends on the relative orientation of the amido substituents. When the SiMe2Ph and Dipp groups are perfectly poised to allow Si–Si bond formation, the activation barrier for ring closure is as low as 11 kJ mol−1 (ESI†). Furthermore, the formation of 19 was found to be highly exergonic (ΔG = −181 kJ mol−1) but less than that calculated for 3. Attempts to find reaction pathways connecting 19 to 3 were met with failure due to the energy demand associated with opening the Si3 ring without an external base (cf. equilibrium VI ↔ VII). This suggests that 19 is a potential thermodynamic sink, and, therefore, a likely candidate for the unidentified reaction product.

Despite several attempts, compound 19 could not be crystallized from the product mixture. Consequently, the synthesis of its SiMe3 analogue 19′ was attempted by carrying out the reduction of {N(SiMe3)Dipp}SiBr3 under highly diluted conditions. The replacement of Ph with Me eliminates the possibility for intramolecular cycloaddition and lowers the kinetic barrier associated with cyclisation, whereas the low concentration increases the likelihood of the formation of 19′ over the Si4 ring I. In good agreement with the above, the cyclotrisilene 19′ was isolated, albeit in minute quantities, allowing its spectroscopic and structural characterisation (Fig. 1 and ESI†). Halogen-substituted cyclopropene analogues are known for germanium,18 but 19′ is the first example of a corresponding silicon species. When the reduction of 2 is carried out under similar conditions used in the synthesis of 19′, an analysis of the reaction mixture by 29Si{1H}-IG-NMR spectroscopy showed two signals in 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio at δ = −5.1 and −10.9 ppm that are comparable to data for 19′, −7.5 and −16.8 ppm, respectively (ESI†). This lends strong support to the proposed mechanism and the key role played by intermediate 10 with its ability to either cyclise, giving 19, or undergo intramolecular cycloaddition, leading to 3.
:1 ratio at δ = −5.1 and −10.9 ppm that are comparable to data for 19′, −7.5 and −16.8 ppm, respectively (ESI†). This lends strong support to the proposed mechanism and the key role played by intermediate 10 with its ability to either cyclise, giving 19, or undergo intramolecular cycloaddition, leading to 3.
In summary, we describe the synthesis and characterisation of the tetracyclic silaheterocycle 3 that was obtained from the reductive debromination of the tribromoamidosilane 2. Experimental and computational analyses of the reaction mechanism implicate that the disilyne 6, generated by the reduction of 2, reacts with the bromosilylene 7 to give the transient intermediate 10. Successive pericyclic reactions coupled by a 1,2-bromine shift result in a two-fold intramolecular C–C bond activation of a single phenyl substituent on one of the SiMe2Ph moieties. The calculated reaction energies agree with the facile formation of 3, while the reactivity of the silylene functionality in 10 corroborates the formation of a second product, the cyclotrisilene 19. Indirect support for the proposed pathway was obtained via complete characterisation of 19′. Efforts to further fine-tune the steric bulk of the amido ligand to allow a high-yielding synthesis of novel halogen-substituted cyclotrisilenes analogous to 19 and 19′ are currently underway.
This project has received funding from the DFG (LI3087/1-1 and Heisenberg Programme LI3087/2-1), the University of Jyväskylä and the European Research Council (ERC) under the European Union's Horizon 2020 Research and Innovation Programme (Grant agreement # 772510 to H. M. T). Computational resources were provided by the Finnish Grid and Cloud Infrastructure (persistent identifier urn:nbn:fi:research-infras-2016072533). We thank J. L. Schwarz for his help with the synthesis of 1 and Prof. W. Uhl and Prof. F. E. Hahn for their generous support.
Conflicts of interest
The authors declare no conflicts of interest.Notes and references
- (a) J. Li, A. Stasch, C. Schenk and C. Jones, Dalton Trans., 2011, 40, 10448 RSC; (b) T. J. Hadlington, J. Li and C. Jones, Can. J. Chem., 2013, 92, 427 CrossRef.
- (a) J. Li, C. Schenk, C. Goedecke, G. Frenking and C. Jones, J. Am. Chem. Soc., 2011, 133, 18622 CrossRef CAS PubMed; (b) T. J. Hadlington and C. Jones, Chem. Commun., 2014, 50, 2321 RSC.
- (a) A. V. Protchenko, K. H. Birjkumar, D. Dange, A. D. Schwarz, D. Vidovic, C. Jones, N. Kaltsoyannis, P. Mountford and S. Aldridge, J. Am. Chem. Soc., 2012, 134, 6500 CrossRef CAS PubMed; (b) A. V. Protchenko, A. D. Schwarz, M. P. Blake, C. Jones, N. Kaltsoyannis, P. Mountford and S. Aldridge, Angew. Chem., Int. Ed., 2013, 52, 568 CrossRef CAS PubMed.
- T. J. Hadlington, M. Hermann, J. Li, G. Frenking and C. Jones, Angew. Chem., Int. Ed., 2013, 52, 10199 CrossRef CAS PubMed.
- A. V. Protchenko, P. Vasko, D. C. Huan Do, J. Hicks, M. Á. Fuentes, C. Jones and S. Aldridge, Angew. Chem., Int. Ed., 2019, 58, 1808 CrossRef CAS PubMed.
- (a) R. D. Rieke, Science, 1989, 246, 1260 CrossRef CAS PubMed; (b) R. D. Rieke, Acc. Chem. Res., 1977, 10, 301 CrossRef CAS.
- J. Keuter, K. Schwedtmann, A. Hepp, K. Bergander, O. Janka, C. Doerenkamp, H. Eckert, C. Mück-Lichtenfeld and F. Lips, Angew. Chem., Int. Ed., 2017, 56, 13866 CrossRef CAS PubMed.
- (a) K. Schwedtmann, A. Hepp, K. Schwedtmann, J. J. Weigand and F. Lips, Eur. J. Inorg. Chem., 2019, 4719 CrossRef CAS; (b) J. Keuter, C. Schwermann, A. Hepp, K. Bergander, J. Droste, M. R. Hansen, N. L. Doltsinis, C. Mück-Lichtenfeld and F. Lips, Chem. Sci., 2020, 11, 5895 RSC.
- K. Suzuki, T. Matsuo, D. Hashzume, H. Fueno, K. Tanaka and K. Tamao, Science, 2011, 331, 1306 CrossRef CAS PubMed.
- J. Wang, R. Liu, W. Ruan, Y. Li., K. C. Mondal, H. W. Roesky and H. Zhu, Organometallics, 2014, 33, 2696 CrossRef CAS.
- (a) K. Suzuki, T. Matsuo, D. Hashizume and K. Tamao, J. Am. Chem. Soc., 2011, 133, 19710 CrossRef CAS PubMed; (b) N. Tokitoh, H. Suzuki, R. Okazaki and K. Ogawa, J. Am. Chem. Soc., 1993, 115, 10428 CrossRef CAS.
- (a) H. Suzuki, N. Tokitoh and R. Okazaki, J. Am. Chem. Soc., 1994, 116, 11572 CrossRef CAS; (b) M. Kira, S. Ishida, T. Iwamoto and C. Kabuto, J. Am. Chem. Soc., 2002, 124, 3830 CrossRef CAS PubMed.
- J. Hicks, P. Vasko, J. M. Goicoechea and S. Aldridge, J. Am. Chem. Soc., 2019, 141, 11000 CrossRef CAS PubMed.
- S. Ishida, T. Tamura and T. Iwamoto, Dalton Trans., 2018, 47, 11317 RSC.
- (a) D. Wendel, A. Porzelt, F. A. D. Herz, D. Sarkar, C. Jandl, S. Inoue and B. Rieger, J. Am. Chem. Soc., 2017, 139, 8134 CrossRef CAS PubMed; (b) L. Zhu, J. Zhang and C. Cui, Inorg. Chem., 2009, 58, 12007 CrossRef PubMed; (c) K. I. Leszczyńska, P. Deglmann, C. Präsang, V. Huch, M. Zimmer, D. Schweinfurth and D. Scheschkewitz, Dalton Trans., 2020, 49, 13218 RSC.
- A. Meller, C. Böker, U. Seebold, D. Bromm, W. Maringgele, A. Heine, R. Herbst-Irmer, E. Pohl, D. Stalke, M. Noltemeyer and G. M. Sheldrick, Chem. Ber., 1991, 124, 1907 CrossRef CAS.
- M. J. Cowley, V. Huch, H. S. Rzepa and D. Scheschkewitz, Nat. Chem., 2013, 5, 876 CrossRef CAS PubMed.
- A. Sekiguchi, Y. Ishida, N. Fukaya and M. Ichinohe, J. Am. Chem. Soc., 2002, 124, 1158 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental and computational details, NMR spectra, crystallographic data, as well as calculated structures and their energies. CCDC 2118008–2118012. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d2cc00298a |
| This journal is © The Royal Society of Chemistry 2022 |