
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Dearomative Michael addition involving enals and 2-nitrobenzofurans realized under NHC-catalysis†
Mateusz
Dyguda‡
a,
Anna
Skrzyńska‡
*a,
Lesław
Sieroń
 b and
Łukasz
Albrecht
*a
b and
Łukasz
Albrecht
*a
aInstitute of Organic Chemistry, Faculty of Chemistry, Lodz University of Technology, Żeromskiego 116, 90-924 Łódź, Poland
bInstitute of General and Ecological Chemistry, Faculty of Chemistry, Lodz University of Technology, Żeromskiego 116, Łódź, 90-924, Poland. E-mail: lukasz.albrecht@p.lodz.pl; anna.skrzynska@p.lodz.pl; Web: http://www.a-teamlab.p.lodz.pl/
First published on 24th March 2022
Abstract
In this manuscript, the first enantioselective dearomative Michael addition between α,β-unsaturated aldehydes and 2-nitrobenzofurans realized under N-heterocyclic carbene activation has been described. The reaction proceeds via addition of homoenolate to Michael acceptors leading to the formation of biologically important heterocycles with high yields and stereoselectivities. Their functionalization potential has been confirmed in selected, diastereoselective transformations.
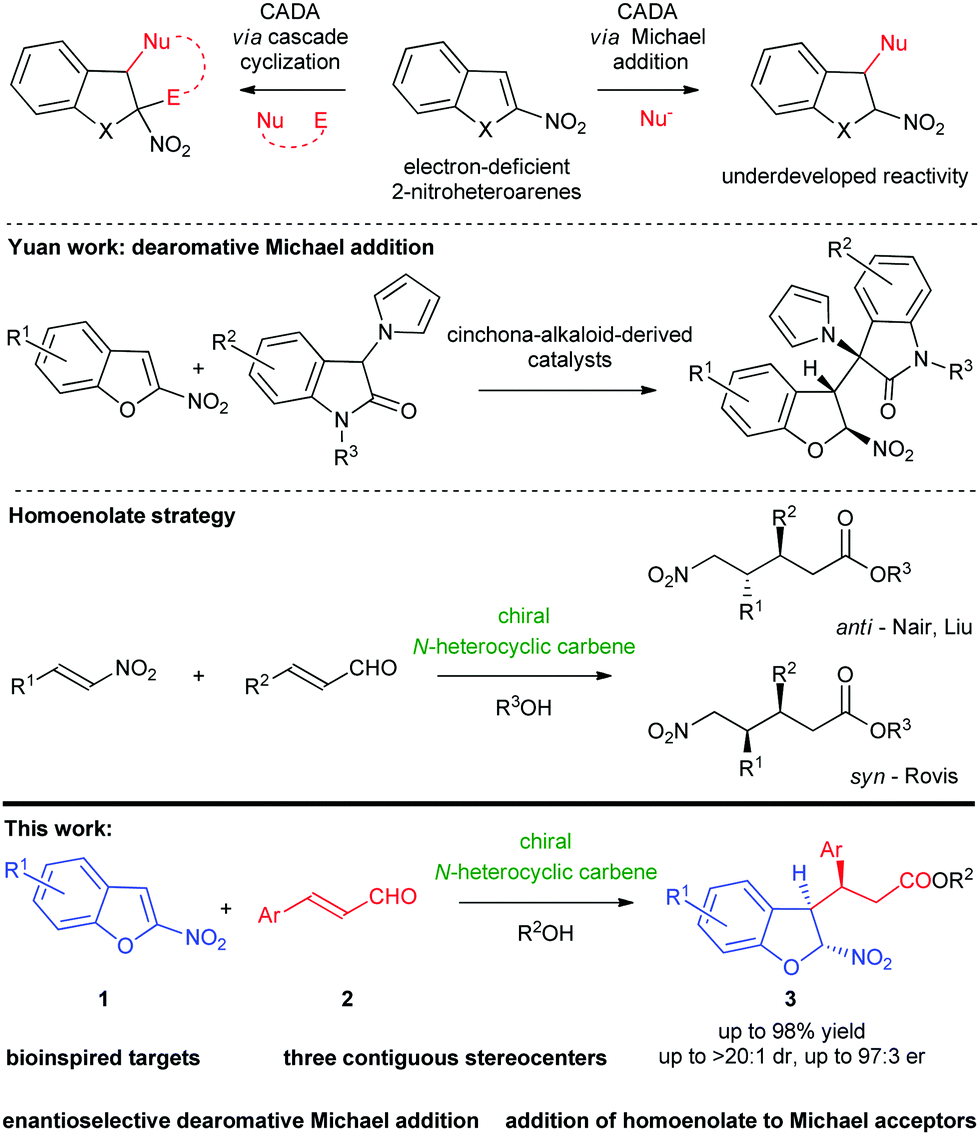
The discovery of new synthetic approaches that allows for the transformation of aromatic systems in a highly selective fashion remains an exciting area of research.1 As a consequence, reactions capable of disturbing the aromatic π-system have received considerable attention. Among different approaches to accomplish this task, catalytic asymmetric dearomatization (CADA) reactions of simple (hetero)aromatic compounds constitute a highly reliable tool for the assembly of enantiomerically enriched polycyclic molecules.2 Many of these reactions are typically focused on the process of dearomatization of electron-rich (hetero)arenes based on their nucleophilic nature.3 Introduction of an appropriate electron-withdrawing substituent in the structure of (hetero)arenes leads to reversal of their reactivity into electron-deficient systems acting as an electrophile susceptible to CADA.4
Recently, important applications of electron-poor nitro-substituted heteroarenes (such as 2- and 3-nitroindoles,5 2-nitrobenzofurans6,7 or 2- and 3-nitrobenzothiophenes8) in dearomative transformations for the construction of fused-heterocycles have appeared in the literature. Due to the immense biological importance of the 2,3-disubstituted-2,3-dihydrobenzoheterocycle motif,9 the development of new strategies for the construction of such chiral oxygen- or sulfur-containing scaffolds has received considerable attention from the organic and medicinal chemistry community. In recent years, 2-nitrobenzofurans and 2-nitrobenzothiophenes have been successfully used as C-2 synthons in various enantioselective dearomative reactions using both transition-metal catalysis6,8a,8b and organocatalysis.7,8c However, all of these transformations proceed in a cascade manner and utilize both the electrophilicity of the starting material and nucleophilicity of the initially formed adduct, thus leading to polycyclic frameworks, mainly (3 + 2)- or (4 + 2)-cycloadducts (Scheme 1, top).6–8 On the other hand, 2-nitrobenzofurans are a class of promising reagents that undergo simple dearomative Michael additions (Scheme 1, top), with such an approach being still undeveloped.
 | ||
| Scheme 1 Design of catalytic asymmetric dearomative reactions of 2-nitrobenzo-heteroarenes. | ||
A survey of the literature revealed only one example of this type of reactivity. In 2019, the Yuan group described the enantioselective reaction of 2-nitrobenzofurans with 3-pyrrolyl-oxindoles in the straightforward construction of chiral 2,3-dihydrobenzofurans (Scheme 1, center).10 Given the lack of simple additions involving 2-nitrobenzofurans and our continuous research activity in catalytic asymmetric dearomative transformations,11 we became interested in the development of the enantioselective CADA Michael reaction between electron-deficient 2-nitrobenzofurans 1 and α,β-unsaturated aldehydes 2. Herein, we report the first organocatalytic dearomative Michael addition realized under NHC catalysis12 leading to the formation of chiral 2,3-disubstituted-2,3-dihydrobenzo-heterocycles 3 (Scheme 1, bottom). The reaction was realized employing homoenolate chemistry that served as a powerful means for the redox functionalization of α,β-unsaturated aldehydes with nitroolefins constituting an important class of electrophiles employed (Scheme 1, center).13
The reaction between 2-nitrobenzofuran 1a and trans-cinnamaldehyde 2a was selected as a model transformation (Table 1). Initially, various readily available chiral NHC catalysts 4 were evaluated in THF at room temperature. It was found that 2-nitrobenzofuran 1a was fully consumed when the reactions were carried out in the presence of the precatalysts 4a,b and d using Cs2CO3 as a base, but 3a was isolated with moderate yields (Table 1, entries 1–2, 4). Aminoindanol-based NHC precatalyst 4c bearing a pentafluorophenyl group showed no catalytic activity in the reaction (Table 1, entry 3). Careful examination of 1H NMR of the crude reaction mixtures indicated that methyl 3-phenylpropanoate was the by-product responsible for the low yields observed. Screening of bases did not lead to significant improvement of the results. However, both Cs2CO3 and triethylamine provided the best reaction outcomes (Table 1, compare entries 4, 7 vs. 5, 6). Subsequently, solvent screening revealed that diethyl ether was the best choice, yielding the desired product 3a without erosion of the enantio- or diastereoselectivity, when reacting with TEA as a base and 4d as a NHC-precatalyst (Table 1, compare entry 11 vs. 7–10). Lowering the reaction temperature to 5 °C led to a substantial increase of the yield with an excellent enantiomeric excess (Table 1, entry 12). It should be noted that under these conditions the formation of methyl 3-phenylpropanoate was not observed in the crude reaction mixture. The decrease of the temperature to −10 °C led to the drop of the diastereoselectivity of the process (Table 1, entry 13). When the catalyst loading 4d was reduced (to 10 mol%), the addition still proceeded smoothly (Table, entry 14). Unfortunately, further reduction of the amount of catalyst (to 5 mol%) suppressed the reaction rate resulting in a lower yield (Table 1, entry 15). Finally, the conditions shown in Table 1, entry 14 were selected as the optimal conditions to investigate the generality of the process. Importantly, the reaction proved to be easily scalable with comparable results obtained on a 20-fold higher scale using only 5 mol% of the precatalyst 4d (Table 1, entry 16).
|
|
|||||
|---|---|---|---|---|---|
| Solvent (preNHC) | Base | Conv. (yield)b [%] | drc | erd | |
| a Reactions performed on a 0.05 mmol scale using 1a (1.0 equiv.), 2a (1.5 equiv.) and the preNHC catalyst 4 (20 mol%) in 0.2 mL of the solvent for 24 h at rt. b Determined by 1H NMR of the crude reaction mixture. In parentheses, the yield of isolated product 3a after column chromatography is given. c Determined by 1H NMR of the crude reaction mixture. d Determined by chiral HPLC. e The reaction was performed at 5 °C. f The reaction was performed at −10 °C. g The reaction was performed using 4d (10 mol%). h The reaction was performed using 4d (5 mol%). i The reaction was performed on a 1 mmol scale for 48 h. DCE – 1,2-dichloroethane. | |||||
| 1 | THF (4a) | Cs2CO3 | >95 (60) | >20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 :1 |
90:10 |
| 2 | THF (4b) | Cs2CO3 | >95 (62) | >20:1 |
15:85 |
| 3 | THF (4c) | Cs2CO3 | <5 | n.d | n.d |
| 4 | THF (4d) | Cs2CO3 | >95 (66) | >20:1 |
92.8 |
| 5 | THF (4d) | K2CO3 | >95 (60) | >20:1 |
90:10 |
| 6 | THF (4d) | DIPEA | >95 (54) | >20:1 |
92:8 |
| 7 | THF (4d) | TEA | >95 (60) | 9:1 |
92:8 |
| 8 | DCE (4d) | Cs2CO3 | <5 | n.d | n.d |
| 9 | 1,4-Dioxane (4d) | Cs2CO3 | <5 | n.d | n.d |
| 10 | Et2O (4d) | Cs2CO3 | >95 (43) | >20:1 |
97:3 |
| 11 | Et2O (4d) | TEA | >95 (76) | >20:1 |
96:4 |
| 12e | Et2O (4d) | TEA | >95 (93) | >20:1 |
96.5:3.5 |
| 13f | Et2O (4d) | TEA | >95 (74) | 16:1 |
96:4 |
| 14eg | Et2O (4d) | TEA | >95 (92) | >20:1 |
96.5:3.5 |
| 15eh | Et2O (4d) | TEA | >95 (74) | >20:1 |
96.5:3.5 |
| 16ehi | Et2O (4d) | TEA | >95 (84) | >20:1 |
96.5:3.5 |

Under the optimized reaction conditions, the scope and limitations of the reaction were carefully explored (Table 2 and Scheme 2). As shown in Table 2, the dearomative Michael reaction was applicable to a wide range of α,β-unsaturated aldehydes 2a–l, affording corresponding products 3a–l in high yields and high diastereo- and enantioselectivity. It was found that the reactivity and stereoselectivity were almost unaffected by the incorporation of various electron-donating substituents at different positions of the aromatic ring in trans-cinnamaldehydes 2b–e (Table 2, products 3b–e). Aldehydes 2f–h with electron-withdrawing substituents in different positions (2-Cl, 3-F and 4-Cl) on the aromatic ring were also all suitable substrates for this reaction, resulting in the formation of the corresponding products 3f–h in high yield and stereoselectivity. Notably, in the case of aldehyde 2i, the reaction proceeded smoothly with 2-nitrobenzofuran 1a, but the desired product 3i was obtained with diminished enantiocontrol. Also trans-cinnamaldehydes 2j and 2k with a double substitution pattern gave access to the corresponding products 3j and 3k in moderate to high enantioselectivity. Finally, the reactivity and stereoselectivity were hardly affected by the incorporation of a heteroaromatic ring in 2l, as 3l was obtained effectively.
|
|
||||
|---|---|---|---|---|
| Entry | 3 (2) | Ar | Yield [%] | er |
| a In the reaction, ent-4d was used as the catalyst and the opposite enantiomer was formed. | ||||
| 1 | 3a (2a) | Ph | 92 | 96.5:3.5 |
| 2 | 3b (2b) | 2-MeOC6H4 | 98 | 93:7 |
| 3 | 3c (2c) | 3-MeOC6H4 | 74 | 94.5:5.5 |
| 4 | 3d (2d) | 4-MeOC6H4 | 79 | 96.5:3.5 |
| 5a | 3e (2e) | 4-MeC6H4 | 78 | 94.5:5.5 |
| 6a | 3f (2f) | 2-ClC6H4 | 74 | 94.5:5.5 |
| 7 | 3g (2g) | 3-FC6H4 | 83 | 95:5 |
| 8 | 3h (2h) | 4-ClC6H4 | 77 | 95:5 |
| 9 | 3i (2i) | 4-NO2C6H4 | 74 | 85:15 |
| 10a | 3j (2j) | 2-Naphthyl | 69 | 80.5:19.5 |
| 11 | 3k (2k) | 2,4-ClC6H3 | 71 | 94:6 |
| 12 | 3l (2l) | 2-Furyl | 87 | 94.5:5.5 |

 | ||
| Scheme 2 NHC-catalyzed dearomative Michael addition with enals and 2-nitrobenzofurans–2-nitrobenzofuran 1 scope. a In the reaction, ent-4d was used as the catalyst and the opposite enantiomer was formed. | ||
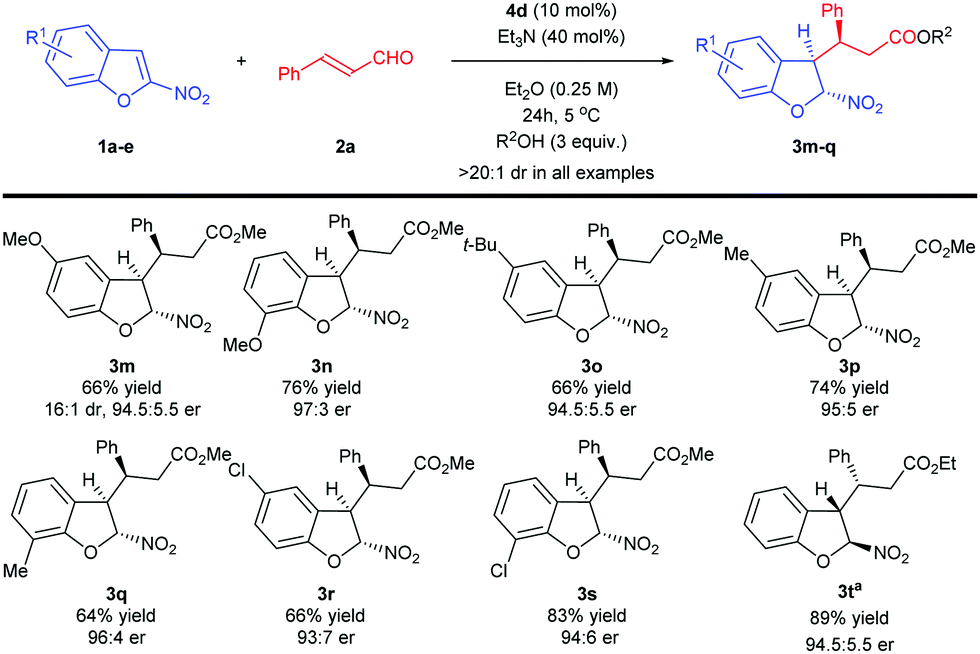
Further exploration of the substrate scope was focused on the utilization of various 2-nitrobenzofurans 1b–h (Scheme 2).
The dearomative Michael reaction was compatible with C5 and C7-substituted acceptors 1 containing groups of different electronic properties, providing products 3m–s in good to high yields and with excellent stereoselection.
Finally, different alcohols, such as ethanol, butanol and sterically hindered tert-butanol were tested. To our delight, ethanol worked well to give 3t in high yield with excellent enantiomeric ratio. Attempts to use other alcohols resulted in suppression of the reactivity (< 5% conversion, products not shown). Similarly, when 2-nitrobenzothiophene, 2-nitroindole, 3-nitroindole and 3-nitrobenzofuran were used, no reaction was observed.
The usefulness of the obtained adducts was demonstrated in selected transformations (Scheme 3). Firstly, the treatment of the optically pure adduct 3a with diisobutylaluminum hydride in THF led to chemoselective reduction of the ester group to give an alcohol 5 in 68% yield without erosion of the diastereoselectivity (Scheme 3, eq. 1). Product 3a was also transformed into the δ-lactam 6via a cascade reaction with sodium borohydride in the presence of nickel chloride in MeOH. Initially, the reduction of the nitro group and lactamization occurred. Subsequently, the reaction involving opening of the dihydrobenzofuran ring and reduction provided 6 as a single diastereoisomer (Scheme 3, eq. 2).
 | ||
| Scheme 3 Diastereoselective transformations of 3a. | ||
The absolute configuration of the products was unequivocally confirmed by the single crystal X-ray analysis of 3j (for details, see the ESI†).14 The stereochemistry of other products was assigned by analogy. Based on the configurational assignments, a possible mechanism of this dearomative Michael reaction was proposed (Scheme 4). It was initiated through the addition of an in situ generated NHC 7 to the α,β-unsaturated aldehyde 2a to give the corresponding Breslow intermediate 8. The Michael acceptor 1a was activated and oriented in space through the H-bonding interaction between the hydroxyl group of 8 and the nitro group in 1a. Simultaneously, π-stacking between the phenyl ring of 8 and aromatic ring of 1a and the steric effect of the chiral motif of the NHC catalyst favored the Re-face attack of the C3-position of 2-nitrobenzofuran by homoenolate 8 in a stereoselective manner. With the formation of adduct 9 accomplished, its protonation and subsequent tautomerization to acyl azolium 10 took place. The esterification of 9 in the presence of a nucleophile furnished the final adduct 3a with (C2R, C3R, C10S) configuration.
 | ||
| Scheme 4 NHC-catalyzed dearomative Michael addition with enals and 2-nitrobenzofurans – mechanistic considerations. | ||
In summary, we have successfully developed the first catalytic asymmetric dearomative transformation between 2-nitrobenzofurans and α,β-unsaturated aldehydes catalyzed by N-heterocyclic carbenes. This process proceeds through the addition of a homoenolate to 2-nitrobenzofurans leading to enantioenriched heterocycles with three contiguous stereocenters with high efficiency and stereoselectivity. The presented work constitutes the unique application of NHC catalysis in the transformation of electron-poor 2-nitrobenzofurans. Further exploration of the catalytic asymmetric dearomatization of electron-deficient heteroarenes is currently underway.
This project was realized within the Własny Fundusz Stypendialny programme (RNN/WFS/9/2021) funds from Lodz University of Technology. This contribution was completed while the first author (MD) was the Doctoral Candidate in the Interdisciplinary Doctoral School of Lodz University of Technology, Poland.
Conflicts of interest
There are no conflicts to declare.Notes and references
- For selected reviews, see: (a) A. R. Pape, K. P. Kaliappan and E. P. Kündig, Chem. Rev., 2000, 100, 2917–2940 CrossRef CAS PubMed; (b) S. P. Roche and J. A. Porco Jr, Angew. Chem., Int. Ed., 2011, 50, 4068–4093 CrossRef CAS PubMed; (c) Q. Ding, X. Zhou and R. Fan, Org. Biomol. Chem., 2014, 12, 4807–4815 RSC; (d) W. C. Wertjes, E. C. Southgate and D. Sarlah, Chem. Soc. Rev., 2018, 47, 7996–8017 RSC.
- For a book, see: (a) S.-L. You, Asymmetric Dearomatization Reactions, Wiley-VCH, Weinheim, 2016. For selected reviews, see: ; (b) W.-T. Wu, L. Zhang and S.-L. You, Chem. Soc. Rev., 2016, 45, 1570–1580 RSC; (c) C. Zheng and S.-L. You, Nat. Prod. Rep., 2019, 36, 1589–1605 RSC; (d) J. An and M. Bandini, Eur. J. Org. Chem., 2020, 4087–4097 CrossRef CAS; (e) Ch. J. Huck and D. Sarlah, Chem, 2020, 6, 1589–1603 CrossRef CAS PubMed; (f) C. Zheng and S.-L. You, ACS Cent. Sci., 2021, 7, 432–444 CrossRef CAS PubMed; (g) B. Baire and V. Nair, Asian J. Org. Chem., 2021, 10, 932–948 CrossRef; (h) Z. Zhang, H. Han, L. Wang, Z. Bu, Y. Xie and Q. Wang, Org. Biomol. Chem., 2021, 19, 3960–3982 RSC; (i) B. Rkein, A. Bigot, L. Birbaum, M. Manneveau, M. De Paolis, J. Legros and I. Chataigner, Chem. Commun., 2021, 57, 27–44 RSC.
- For selected reviews, see: (a) J. Nan, Z. Zuo, L. Luo, L. Bai, H. Zheng, Y. Yuan, J. Liu, X. Luan and Y. Wang, J. Am. Chem. Soc., 2013, 135, 17306–17309 CrossRef CAS PubMed . For recently examples, see: ; (b) P. Wang, J. Wang, L. Wang, D. Li, K. Wang, Y. Liu, H. Zhu, X. Liu, D. Yang and R. Wang, Adv. Synth. Catal., 2018, 360, 401–405 CrossRef CAS; (c) X. Liu, J. Zhang, L. Bai, L. Wang, D. Yang and R. Wang, Chem. Sci., 2020, 11, 671–676 RSC; (d) M.-L. Han, W. Huang, Y.-W. Liu, M. Liu, H. Xu, H. Xiong and H.-X. Dai, Org. Lett., 2021, 23, 172–177 CrossRef CAS PubMed.
- For selected examples, see: (a) I. Chataigner and S. R. Piettre, Org. Lett., 2007, 9, 4159–4162 CrossRef CAS PubMed; (b) S. Lee, I. Chataigner and R. S. Piettre, Angew. Chem., Int. Ed., 2010, 50, 472–476 CrossRef PubMed; (c) B. M. Trost, V. Ehmke, B. M. O’Keefe and D. A. Bringley, J. Am. Chem. Soc., 2014, 136, 8213–8216 CrossRef CAS PubMed.
- For the selected examples about the application of nitroindoles in asymmetric reactions, see: (a) Y. Li, F. Tur, R. P. Nielsen, H. Jiang, F. Jensen and K. A. Jørgensen, Angew. Chem., Int. Ed., 2016, 55, 1020–1024 CrossRef CAS PubMed; (b) D.-F. Yue, J.-Q. Zhao, X.-Z. Chen, Y. Zhou, X.-M. Zhang, X. Y. Xu and W.-C. Yuan, Org. Lett., 2017, 19, 4508–4511 CrossRef CAS PubMed; (c) J.-Q. Zhao, X.-J. Zhou, Y.-Z. Chen, X.-Y. Xu, X.-M. Zhang and W.-C. Yuan, Adv. Synth. Catal., 2018, 360, 2482–2487 CrossRef CAS; (d) H.-M. Wang, J.-Y. Zhang, Y.-S. Tu and J.-L. Zhang, Angew. Chem., Int. Ed., 2019, 58, 5422–5426 CrossRef CAS PubMed; (e) K.-Z. Li, T. P. Goncalves, K.-W. Huang and Y. Lu, Angew. Chem., Int. Ed., 2019, 58, 5427–5431 CrossRef CAS PubMed.
- For the selected examples about the application of nitrobenzofuranes in transition-metal catalysis, see: (a) J.-Q. Zhao, X.-J. Zhou, Y. Zhou, X.-Y. Xu, X.-M. Zhang and W.-C. Yuan, Org. Lett., 2018, 20, 909–912 CrossRef CAS PubMed; (b) Q. Cheng, H.-J. Zhang, W.-J. Yue and S.-L. Yo, Chem, 2017, 3, 428–436 CrossRef CAS.
- For the selected examples about the application of nitrobenzofurans in organocatalytic reactions, see: (a) X.-J. Zhou, J.-Q. Zhao, X.-M. Chen, J.-R. Zhuo, Y.-P. Zhang, Y.-Z. Chen, X.-M. Zhang, X.-Y. Xu and W.-C. Yuan, J. Org. Chem., 2019, 84, 4381–4391 CrossRef CAS PubMed; (b) X.-H. Yang, J.-P. Li, D.-C. Wang, M.-S. Xie, G.-R. Qu and H.-M. Guo, Chem. Commun., 2019, 55, 9144–9147 RSC.
- For the selected examples about the application of nitrobenzothiophenes in asymmetric reactions, see: (a) Q. Cheng, F. Zhang, Y. Cai, Y.-L. Guo and S.-L. You, Angew. Chem., Int. Ed., 2018, 57, 2134–2138 CrossRef CAS PubMed; (b) D.-F. Yue, J.-Q. Zhao, Y.-Z. Chen, X.-M. Zhang, X.-Y. Xu and W.-C. Yuan, Adv. Synth. Catal., 2018, 360, 1420–1425 CrossRef CAS; (c) X.-M. Chen, C.-W. Lei, D.-F. Yue, J.-Q. Zhao, Z.-H. Wang, X.-M. Zhang, X.-Y. Xu and W.-C. Yuan, Org. Lett., 2019, 21, 5452–5456 CrossRef CAS PubMed.
- For selected review, see: (a) Z. Chen, M. Pitchakuntla and Y. Jia, Nat. Prod. Rep., 2019, 36, 666–690 RSC ; For selected examples, see: ; (b) T. Hayashi and R. H. Thomson, Phytochemistry, 1975, 14, 1085–1087 CrossRef CAS; (c) Y. Mimaki, A. Kameyama, Y. Sashida, Y. Miyata and A. Fujii, Chem. Pharm. Bull., 1995, 43, 893–895 CrossRef CAS PubMed.
- Z.-Z. Ge, L. Yang, Y. You, Z.-H. Wang, K.-X. Xie, M.-Q. Zhou, J.-Q. Zhao and W.-C. Yuan, Chem. Commun., 2020, 56, 2586–2589 RSC.
- (a) A. Skrzyńska, A. Przydacz and Ł. Albrecht, Org. Lett., 2015, 17, 5682–5685 CrossRef PubMed; (b) X. Y. Gao, R. J. Yan, B. X. Xiao, W. Du, Ł. Albrecht and Y.-C. Chen, Org. Lett., 2019, 21, 9628–9632 CrossRef CAS PubMed; (c) J. Bojanowski, A. Skrzyńska and A. Albrecht, Asian J. Org. Chem., 2019, 8, 844–848 CrossRef CAS; (d) A. Przydacz, M. Dyguda, A. Topolska, A. Skrzyńska, C.-J. Xu, Y.-C. Chen and Ł. Albrecht, Org. Biomol. Chem., 2020, 18, 5816–5821 RSC; (e) A. Skrzyńska, S. Frankowski, A. Topolska, M. Dyguda, X.-Y. Gao, Ch.-J. Xu, Y.-C. Chen and Ł. Albrecht, Chem. Commun., 2021, 57, 1667–1670 RSC; (f) M. Saktura, S. Frankowski, B. Joachim and Ł. Albrecht, Synthesis, 2021, 309–317 CAS; (g) M. Saktura, A. Skrzyńska, S. Frankowski, S. Wódka and Ł. Albrecht, Molecules, 2021, 26, 4992–5002 CrossRef CAS PubMed.
- For a book, see: (a) A. T. Biju, N-Heterocyclic carbenes in Organocatalysis, Wiley-VCH, Weinheim, 2019. For selected reviews, see: ; (b) D. M. Flanigan, F. Romanov-Michailidis, N. A. White and T. Rovis, Chem. Rev., 2015, 115, 9307–9387 CrossRef CAS PubMed; (c) M. Zhao, Y.-T. Zhang, J. Chen and L. Zhou, Asian J. Org. Chem., 2018, 7, 54–69 CrossRef CAS.
- For selected reviews on homoenolate chemistry realized under NHC catalysis: (a) V. Nair, R. S. Menon, A. T. Biju, C. R. Sinu, R. R. Paul, A. Jose and A. Sreekumar, Chem. Soc. Rev., 2011, 40, 5336–5346 RSC; (b) R. S. Menon, A. T. Biju and V. Nair, Chem. Soc. Rev., 2015, 44, 5040–5052 RSC; (c) B. Maij, Asian J. Org. Chem., 2018, 7, 70–84 CrossRef . For examples involving nitroolefins as electrophiles, see: ; (d) V. Nair, C. R. Sinu, B. P. Babu, V. Varghese, A. Jose and E. Suresh, Org. Lett., 2009, 11, 5570–5573 CrossRef CAS PubMed; (e) B. Maji, L. Ji, S. Wang, S. Vedachalam, R. Ganguly and X.-W. Liu, Angew. Chem., Int. Ed., 2012, 51, 8276–8280 CrossRef CAS PubMed; (f) N. A. White, D. A. DiRocco and T. Rovis, J. Am. Chem. Soc., 2013, 135, 8504–8507 CrossRef CAS PubMed.
- CCDC 2105519 contains the supplementary crystallographic data for this paper†.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 2105519. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d2cc00294a |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2022 |