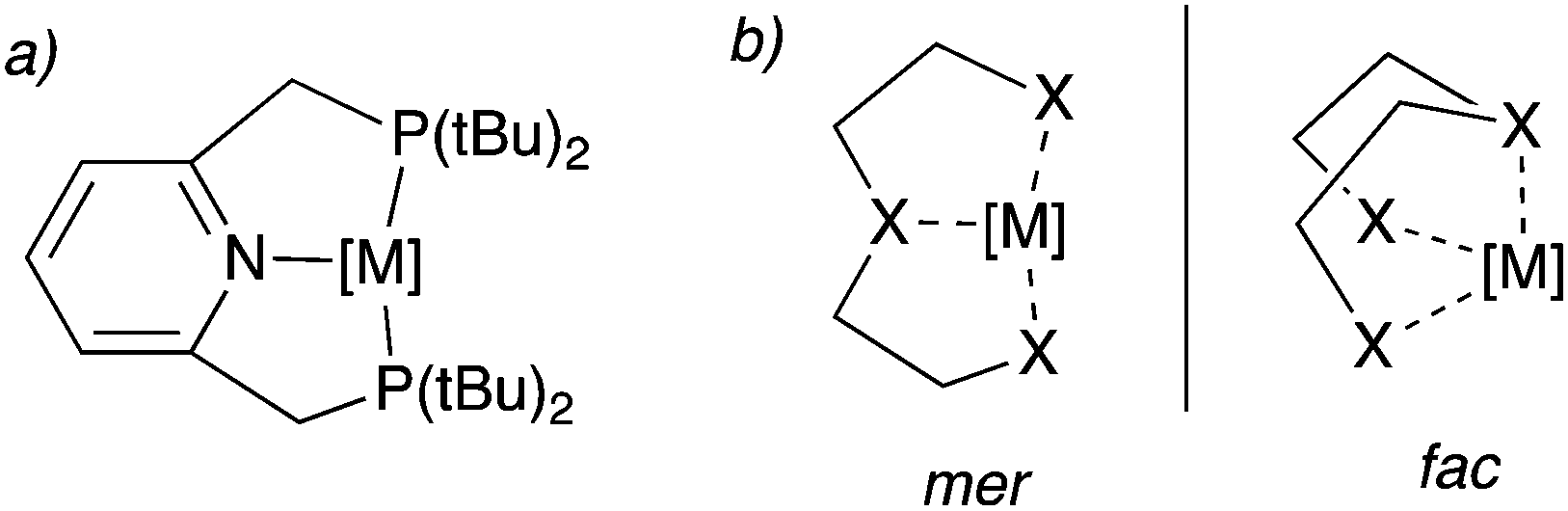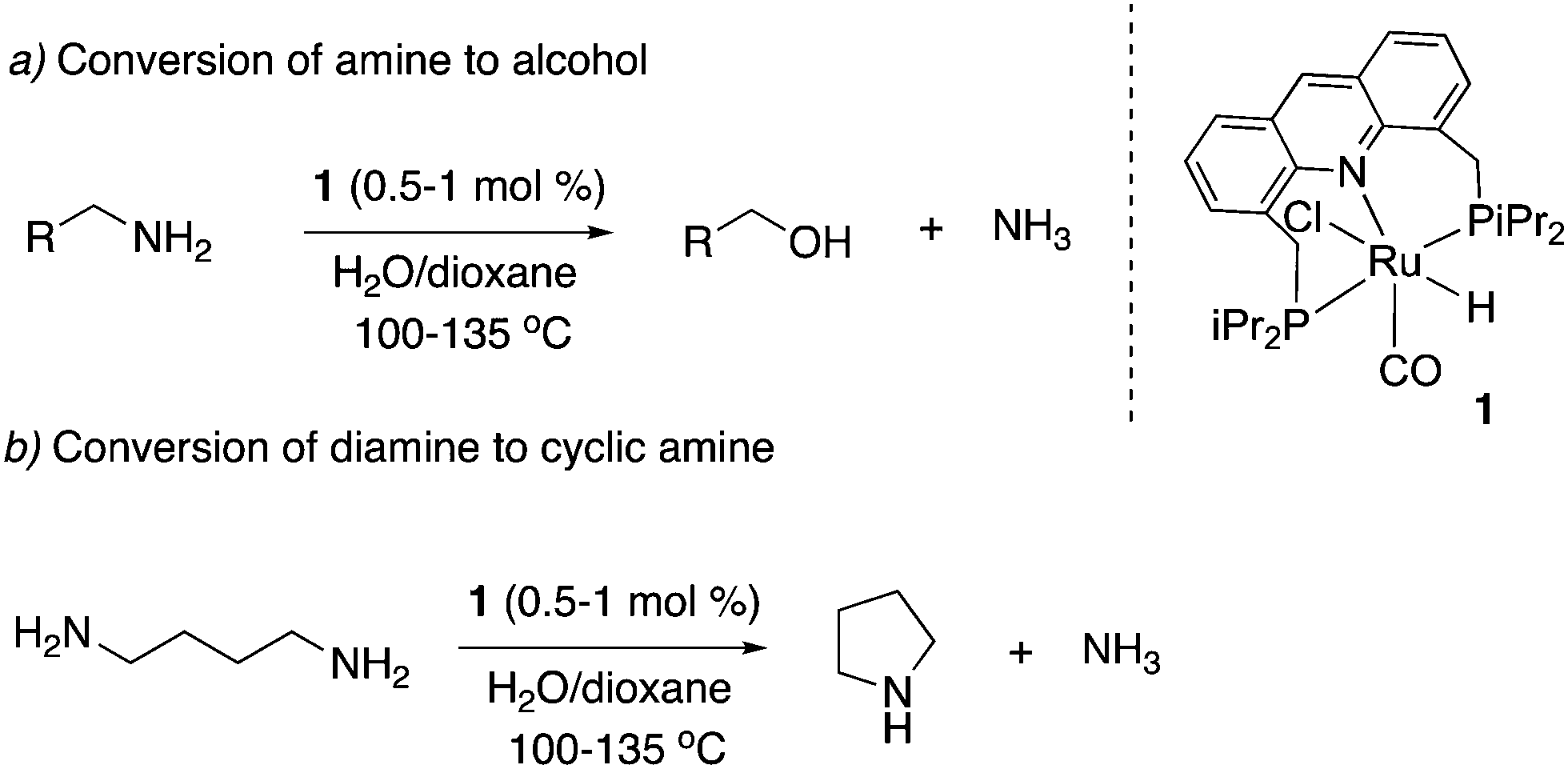DOI:
10.1039/D2CC00247G
(Feature Article)
Chem. Commun., 2022,
58, 3731-3746
Sustainable catalysis with fluxional acridine-based PNP pincer complexes
Received
13th January 2022
, Accepted 23rd February 2022
First published on 23rd February 2022
Abstract
Because of the widespread use of fossil fuels and the resulting global warming, development of sustainable catalytic transformations is now more important than ever to obtain our desired fuels and building materials with the least carbon footprint and waste production. Many sustainable (de)hydrogenation reactions, including CO2 reduction, H2 carrier systems, and others, have been reported using molecular pincer complexes. A specific subset of pincer complexes containing a central acridine donor with flanking CH2PR2 ligands, known as acridine-based PNP pincer complexes, exhibit special reactivities that are not imitable by other PNP pincer complexes such as pyridine-based or (R2PCH2CH2)2NH type ligands. The goal of this article is to highlight the unique reactivities of acridine-based complexes and then investigate how these reactivities allow these complexes to catalyse many sustainable reactions that traditional pincer complexes cannot catalyse. To that end, we will initially go over the synthesis and structural features of acridine complexes, such as the labile coordination of the central N donor and the observed fac–mer fluxionality. Following that, distinct reactivity patterns of acridine-based complexes including their reactivity with acids and water will be discussed. Finally, we will discuss the reaction systems that have been developed with acridine complexes thus far, including the notable selective transformations of primary alcohols to primary amines using ammonia, N-heteroaromatic synthesis from alcohols and ammonia, oxidation reactions with water with H2 liberation, development of H2 carrier systems, and others, and conclude the article with future possible directions. We hope that the systemic study presented here will aid researchers in developing further sustainable reactions based on the unique acridine-based pincer complexes.

Sayan Kar
| Sayan Kar received his Integrated MSc degree (Chemistry) at Indian Institute of Technology Kanpur (India) in 2015. He received his PhD in 2019 at University of Southern California (Los Angeles, USA) with Prof. G. K. Surya Prakash as PhD advisor. Subsequently, he joined the research group of Prof. David Milstein at Weizmann Institute of Science (Israel) as a postdoctoral researcher, focusing of the development of sustainable catalytic reactions with transition metal pincer complexes. He is currently a visiting postdoctoral researcher in the group of Prof. Erwin Reisner at University of Cambridge (United Kingdom). |

David Milstein
| David Milstein is the Israel Matz Professor of Chemistry at the Weizmann Institute of Science. He received his PhD degree with Prof. Blum at the Hebrew University in 1976 and performed postdoctoral research at Colorado State University with Prof. Stille. In 1979 he joined DuPont Company's CR&D where he became a Group Leader, and in 1986 he moved to the Weizmann Institute, where he headed the Department of Organic Chemistry in 1996–2005. In 2000, he founded and became Head of the Kimmel Center for Molecular Design at the Weizmann Institute until 2017. He has received several awards, including the Israel Prize in 2012. He is a member of the Israel Academy of Sciences and Humanities; US National Academy of Science; German National Academy of Sciences-Leopoldina, European Academy of Sciences, and Foreign Member of the Royal Society (ForMemRS, UK). |
1. Introduction
Over the last two decades, pincer complexes have gained significant attraction in catalysis.1–6 Traditionally, pincer complexes are defined as complexes bearing a tridentate ligand (the pincer ligand), which has a clear preference for meridional binding geometry, meaning the three donor atoms of the pincer ligand preferentially bind in three adjacent coordination sites on the same plane (Fig. 1a). However, later definitions also allow for a mer–fac fluxionality (Fig. 1b). The tri-coordinating nature of the pincer ligand provides high stability to the complex and provides structural conformation, which assists in their activity for many catalytic reactions even at high reaction temperatures (>150 °C). Initial pincer complexes with tridentate ligands were reported in the 1970s, and Van Koten coined the term pincer complexes.7–9 Since then, many pincer complexes have been synthesized and applied in catalysis, which has been masterfully and categorically summarized and highlighted in recent reports.3–6,10
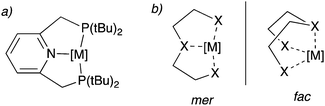 |
| | Fig. 1 (a) a typical pincer complex with pyridine base PNPtBu pincer ligand. (b) meridional (mer) and facial (fac) binding mode of the pincer ligands. | |
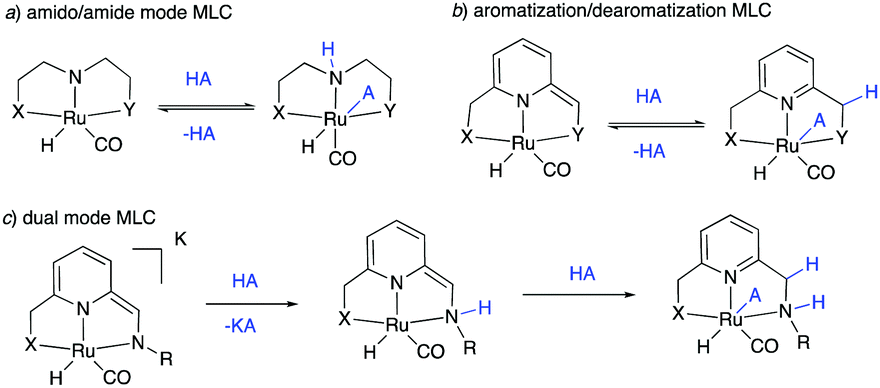
The field of homogeneous sustainable catalysis has seen tremendous progress in recent decades.11–15 Among the many directions in this broad area, seminal works were done in H2 storage,12,16–19 CO2 hydrogenation,20–24 methanol reforming,25–27 waste recycling,28 sustainable organic synthesis, and others. Pincer complexes have played an instrumental part in developing these reactions, often due to their unique ability to reversibly activate bonds such as H–H, N–H, O–H via metal–ligand cooperation (MLC). In this regard, two common classical modes among redox innocent MLCs must be mentioned—one through the formation of metal amido/amine bond (Fig. 2a)29–32 and the other by aromatization–dearomatization of the central pyridine ring (Fig. 2b),33–37 with recent studies reporting complexes featuring both kinds of MLC (Fig. 2c).38–41 Majority of initial reports in this area employed noble metal-based complexes for catalysis; however, over the last decade, first-row transition metal-based complexes—especially manganese, iron and cobalt—have also been intensively studied. Several excellent reviews exist in this promising research area to which the reader can refer to for detailed information.42–46
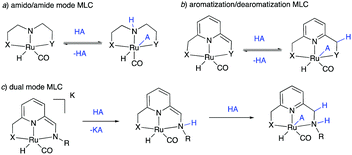 |
| | Fig. 2 Various types of redox innocent metal–ligand cooperation. (A) Amido-amine mode, (b) aromatization–dearomatization mode, and (c) dual-mode. | |
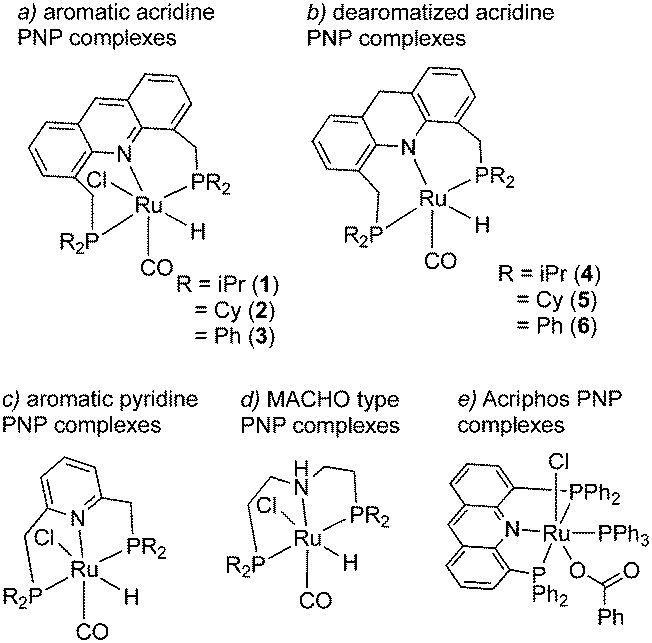
The topic of this feature article is a set of pincer complexes, namely acridine-based PNP-pincer complexes (Fig. 3). The central backbone of the ligand features an acridine ring, as opposed to the more commonly employed pyridine or benzene backbone.47–49 In addition, the P atom is not directly connected to the ring, generating two six membered metallacycles upon coordination, unlike the acriphos (4,5-bis(diphenylphosphino)acridine) ligand (Fig. 3e),50,51 which makes the whole structure flexible. This increased flexibility, in turn, imparts special reactivity on the system, often by allowing temporarily access the fac isomer, as we will later see, which is not possible with other pincer complexes featuring rigid ligand frameworks. Over the last decade, many important sustainable catalytic reactions have been reported with these kinds of complexes, which remain elusive with other pincer complexes. The subsequent sections will briefly discuss their synthesis and structural properties, followed by their particular reactivity compared to other complexes. Subsequently, we will go through the catalytic reactions achieved by employing these complexes as catalysts and afterward conclude with the future possible directions regarding further development.
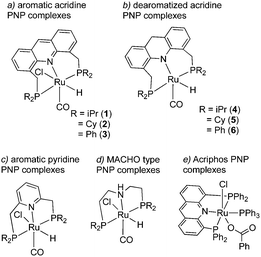 |
| | Fig. 3 Different pincer complexes. | |
2. Synthesis and structure of the complexes
2.1. Synthesis of the ligand and complexation
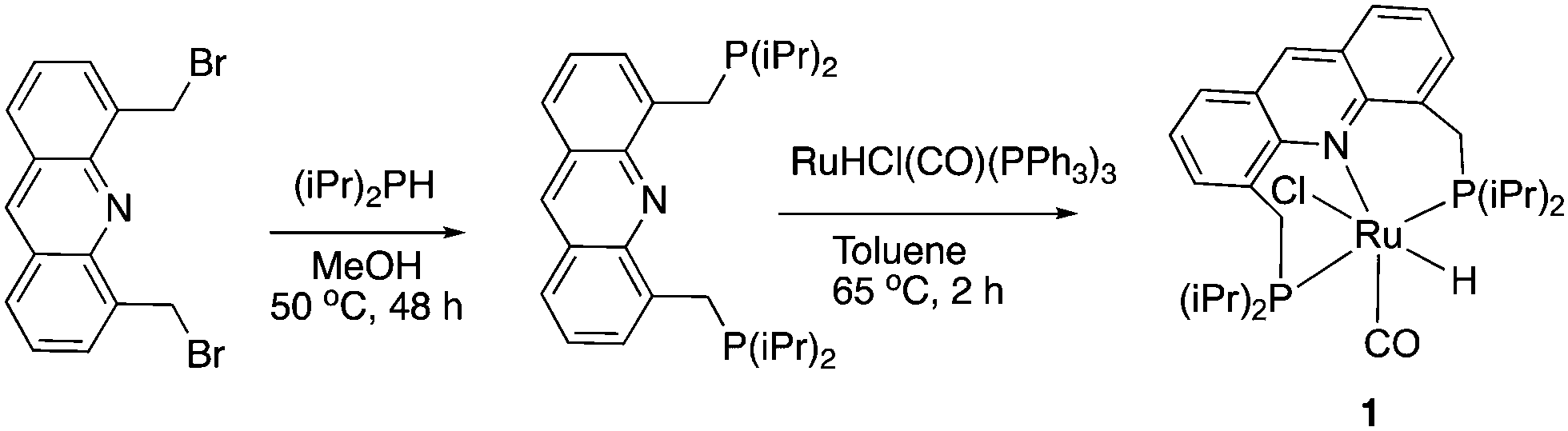
Our group reported the initial example of an acridine PNP complex in 2008, the Ru(II) complex 1 (Fig. 4), and employed it for the catalytic amination of primary alcohols to primary amine using ammonia (more on that later).52 The acridine-based pincer ligand was synthesized by reaction of 4,5-dibromomethyl acridine with PHiPr2 in methanol (Fig. 4) followed by ligand complexation using RuHCl(CO)(PPh3)3, forming complex 1 in quantitative yield as a bronze-coloured powder. Among the characteristic NMR peaks, the P signal appears at 69.35 ppm in the 31P NMR, and in 1H NMR spectra, the hydride peak is seen around −16 ppm as a triplet in C6D6, whereas the 9H acridine ring proton is observed at 8.2 ppm as a singlet. The complex is air-stable for several months.
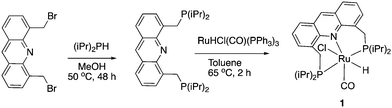 |
| | Fig. 4 Synthesis of complex 1. | |
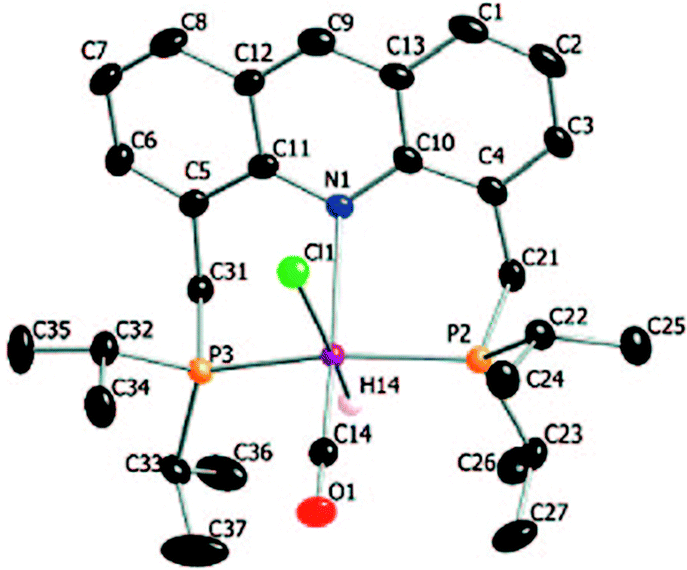
2.2. X-Ray structure
The X-ray structure of complex 1 reveals a distorted octahedral geometry around the ruthenium centre (Fig. 5).53 An elongated Ru–N bond distance was measured (2.479 Å), as compared to 2.103 Å in the pyridine-based systems, suggesting potential hemilability of the binding. Further, the aromaticity of the acridine ring decreased upon complexation, the acridine ring adopting a boat shape with a dihedral angle of 167.6°, likely due to geometrical constraints in coordination. The carbonyl ligand is located trans to the coordinating N atom with the other two axial positions occupied by the hydride and chloride ligands. The acridine complex analog 2, Ru-AcrPNP(Cy) displays similar structural features.54
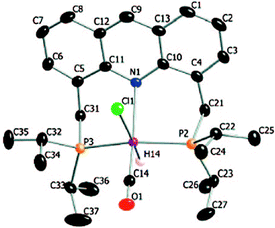 |
| | Fig. 5 Crystal structure of complex 1. Adapted with permission from ref. 53. Copyright 2009 American Chemical Society. | |
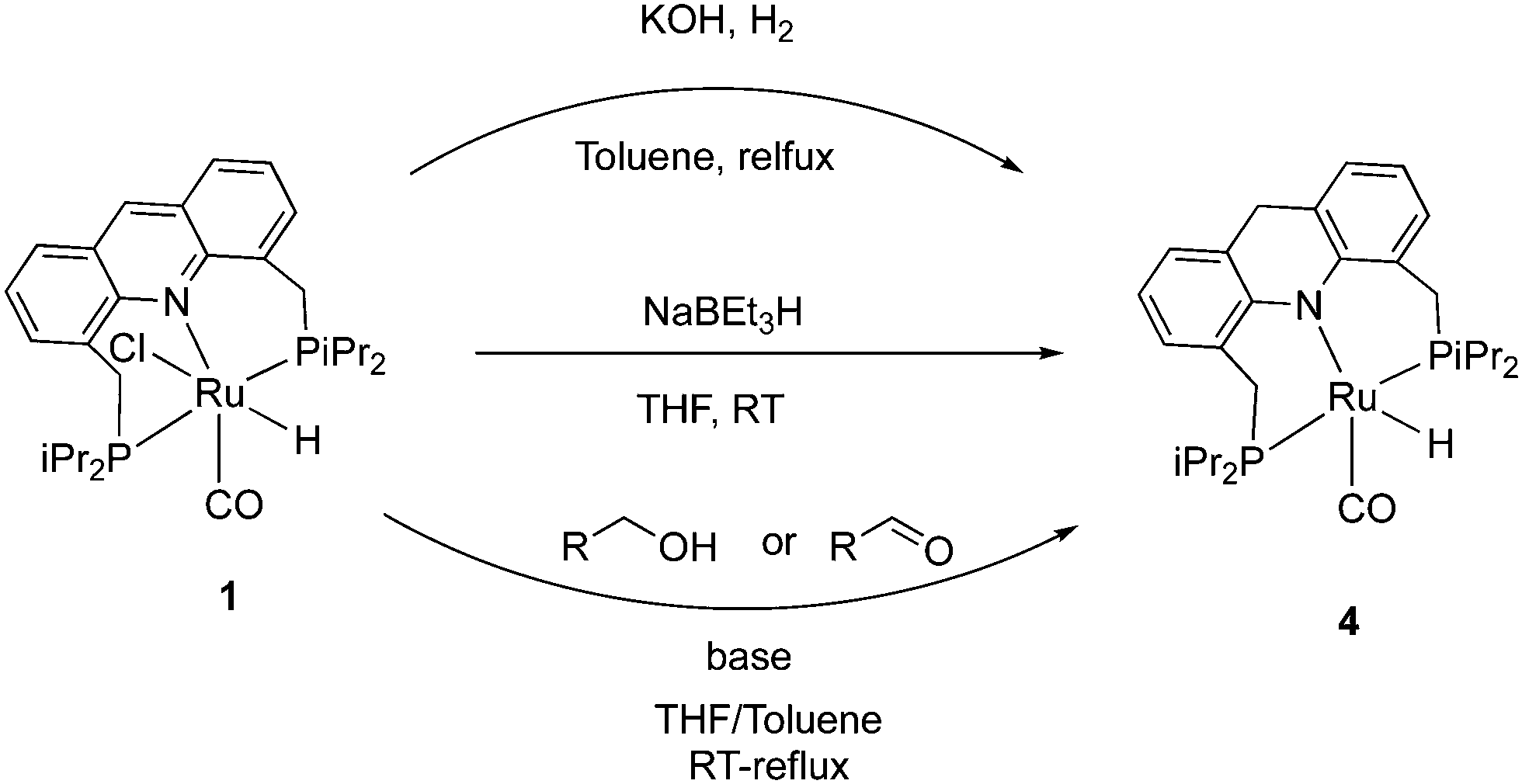
2.3. Dearomatization
The 9CH position of the acridine ring in complex 1 is electrophilic and as a result, it can readily form the corresponding dearomatized acridine PNP complex 4 under various conditions. The dearomatized complex often acts as the real catalyst in systems employing the aromatic complex 1 as pre-catalyst. Among different procedures to access 4 from 1, using H2 gas with KOH at reflux conditions produced the dearomatized complex in high yields (Fig. 6).55 Using D2 instead of H2, formation of C-D (position 9) and Ru–D was observed, suggesting a D2 activation by “long-range” metal–ligand cooperation pathway for the generation of the dearomatized complex. The dearomatized complex can also be accessed by other methods, including addition of one equiv. of the superhydride NaBEt3H to a solution of complex 1 (THF, 30 min, RT),56 or by the reaction of complex 1 with alcohol and 1 equiv. base (KH) at room temperature where the benzyl alcohol is oxidized to benzaldehyde during the process (Fig. 6).54 Among characteristic features in NMR, the 31P peak of the dearomatized complex appears at 74.5 ppm in C6D6, whereas the hydride appears as a triplet at −21.8 ppm in the 1H{31P} NMR due to coupling with the P atoms. The dearomatization is further reflected in the observed aliphatic 9-CH2 protons (AB system) in the 1H NMR around 3.8 ppm in C6D6.
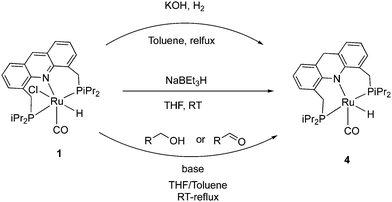 |
| | Fig. 6 Reported procedures to access dearomatized acridine complex 4 from 1. | |
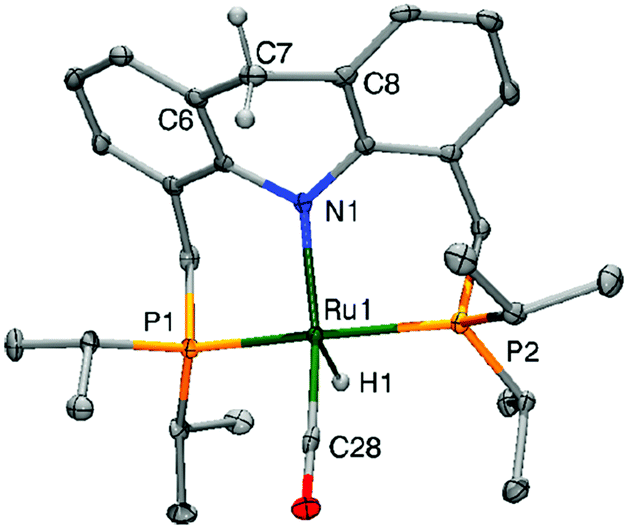
2.4. Structure of the dearomatized complex
The crystal structure of the dearomatized Ru–dAcrPNP(iPr) complex 4 was reported by our group in 2010, showing a distorted trigonal bipyramidal geometry at the ruthenium centre (Fig. 7).55 The Ru–N bond is dramatically shortened upon dearomatization to 2.171 Å from 2.479 Å in complex 1, reflecting the newly formed Ru–amido bond. An axial linear N–Ru–C arrangement is formed, and the equatorial P–Ru–P angle is 160.55°. Notably, the ligand acridine ring of the dearomatized complex is similarly boat-shaped, as in the aromatic 1, with the acridine moiety slightly tilted toward the ruthenium centre.
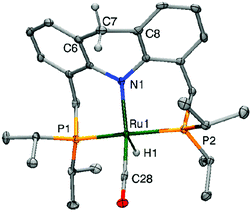 |
| | Fig. 7 X-Ray structure of the dearomatized complex 4. Adapted with permission from ref. 57. Copyright 2015 American Chemical Society. | |
2.5. Irreversibility
The dearomatization of the complex is seemingly irreversible in the acridine systems, and the synthesis of aromatic acridine complexes from dearomatized complexes has not been achieved thus far. This is in stark contrast to the pyridine-based PNP and PNN type pincer complexes, where the reversible formation of aromatized and dearomatized complexes play an instrumental role in bond activation and catalysis (Fig. 2).
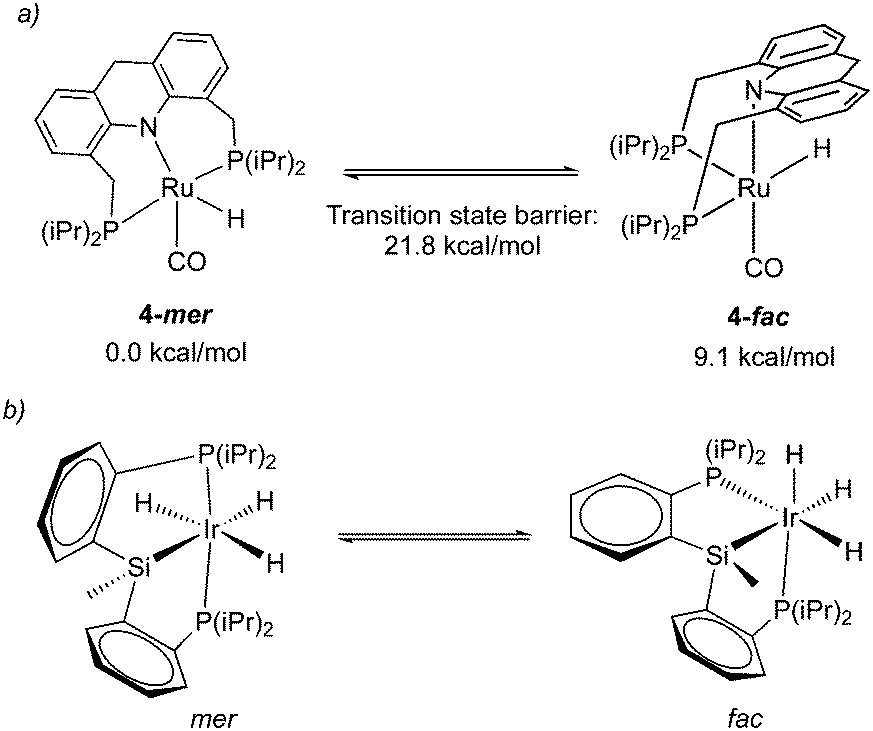
2.6. Fluxionality
While the dearomatized Ru–acridine complex adopts mer coordination as observed by the NMR spectrum and crystal structure, it is surmised to be fluxional to readily access the fac isomer. This has been proposed due to the observation of several dearomatized complexes with fac PNP coordination (the carboxylate complex, thiolate complex) that can be readily synthesized from the parent complex 4 upon the addition of substrates. Further, one of such derivatives, a thiol adduct complex, is also observed in NMR in both fac and mer form, with the fac isomer slowly converting to the more thermodynamically stable mer isomer over the course of two hours.58 Density functional (DFT) calculations indicate that the free energy of the fac isomer is 9.1 kcal mol−1 higher than that of the mer isomer, with the fac/mer conversion activation barrier being 21.8 kcal mol−1 (Fig. 8a).57 While the significant energy difference means that in solution, the fac isomer is not observed by NMR, the small activation barrier suggests that it can nonetheless be accessed, quite possibly even under ambient temperature. In a very recent report from our group, the free energy difference between the two isomers in the dAcr(Ph) system was calculated to be even lower (5.6 kcal mol−1).59 The fluxionality is further reflected in the NMR peak broadening reported for the Ru–dAcrPNP(Ph) system. This type of fluxionality between the fac and mer isomers, while being rare, was also observed with other pincer systems (Fig. 8b).60
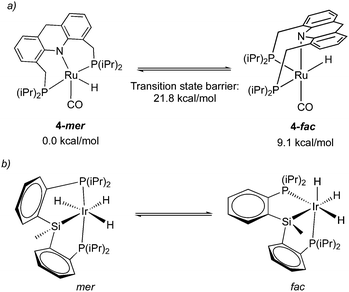 |
| | Fig. 8 (a) fluxionality in complex 4, energies taken from ref. 57. (b) fac–mer isomerization in an iridium pincer complex. | |
3. Special reactivity of the acridine based PNP complexes
Owing to its unique structural properties, the acridine-based PNP complexes show unique reactivities, which are not imitable by other pincer complexes. Some of these properties are detailed below.
(a) Reactivity with acid
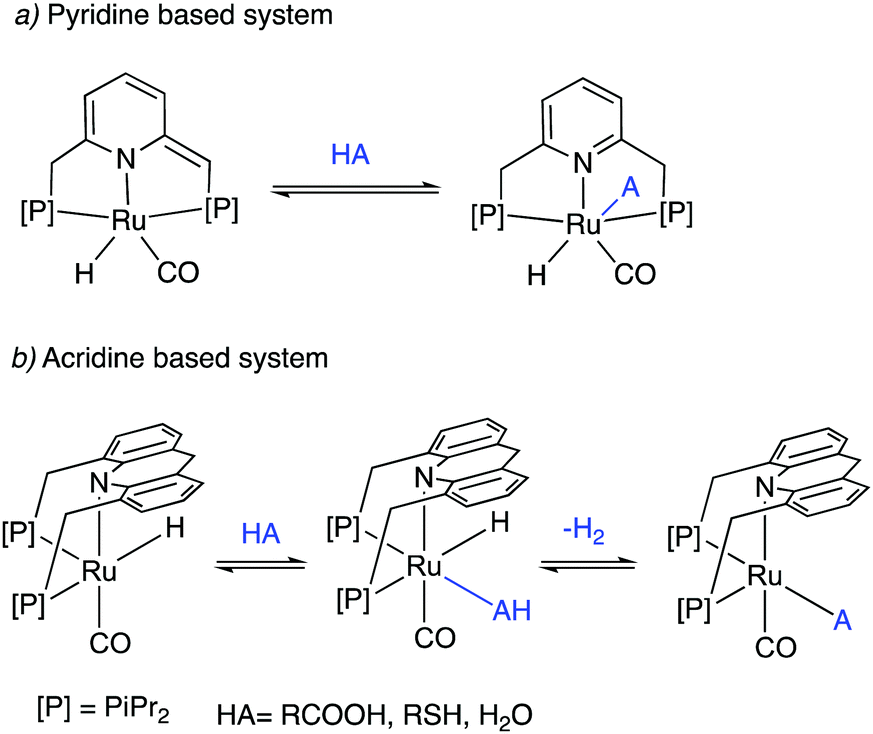
Dearomatized pincer complexes preferring rigid meridional coordination are deactivated in the presence of acids, by the protonation of the ligand arm, forming of deactivating aromatic/amine complexes (Fig. 9a). On the other hand, the donor nitrogen atom in the 9-H acridine (dearomatized) system is only weakly basic due to its conjugation with two adjacent benzene rings, making these complexes acid resistant. Further, these complexes can also liberate H2 from acids through a unique pathway involving the fac isomer, which is often an elementary step in catalysis by these complexes (Fig. 9b).
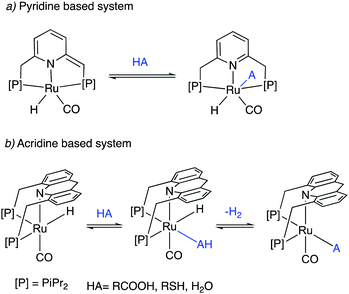 |
| | Fig. 9 The reaction of acid with (a) dearomatized pincer complexes, and (b) with acridine-based dearomatized PNP complexes. | |
(b) Water resistance and cooperation
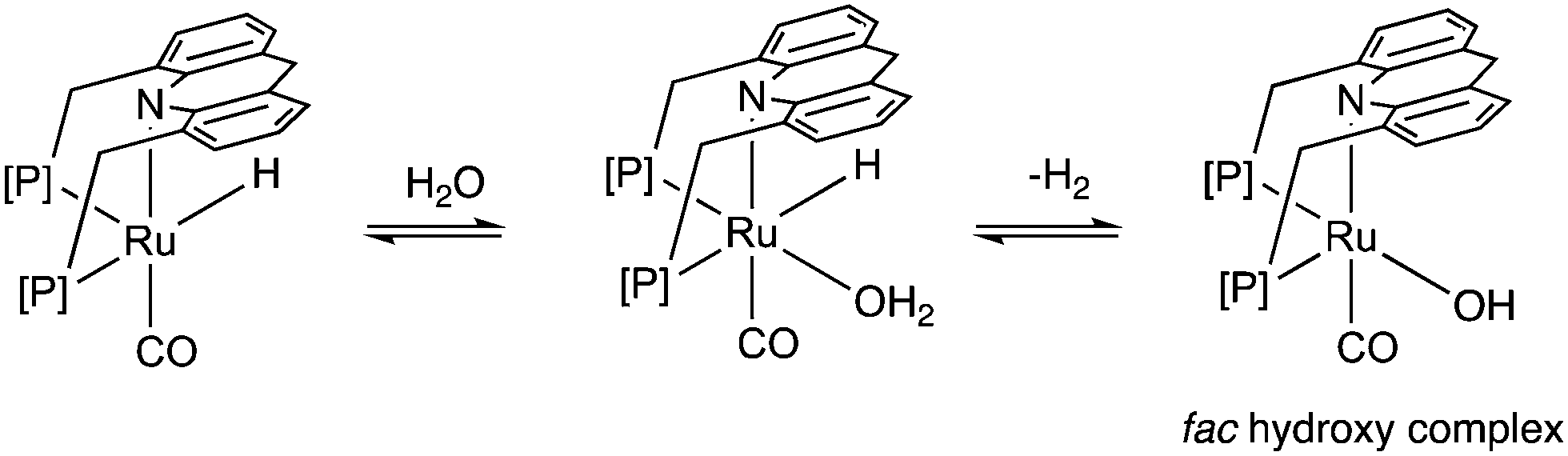
Similar to acids, water also diminishes the activities of dearomatized pincer complexes through the formation of hydroxy complexes. On the other hand, the acridine-based complexes are active in catalysis even in the presence of large amounts of water. This has led to the recent development of many new sustainable reactions, using these complexes as catalysts, where water not only is a (co)solvent but also an oxidant during the process with liberation of H2. Interestingly, a recent study shows that the dearomatized acridine complexes not only can withstand considerable amounts of water, but the presence of water also facilitates its amine dehydrogenation activity through the formation of a fac hydroxy intermediate complex (Fig. 10).56 These properties make the acridine-based complexes special for developing water-based oxidation reactions.
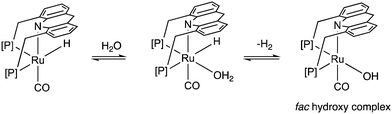 |
| | Fig. 10 Reactivity of dearomatized acridine complexes with water. | |
(c) High stability
The tridentate PNP ligand provides the acridine complex with high stability, similar to other pincer complexes. It is reportedly stable even at high temperatures of 160 °C and above for an extended period. Further, the acridine PNP(iPr) complex has been functional in neat formic acid for over two months.
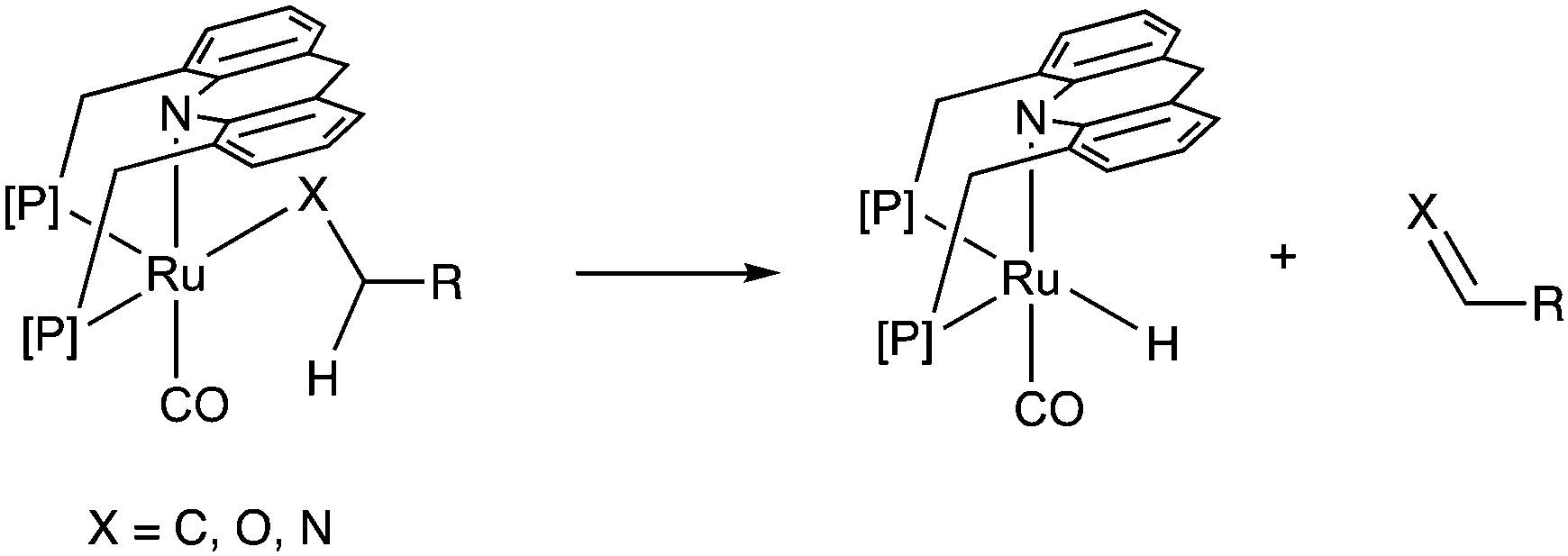
(d) Beta hydride elimination
Adoption of the fac isomer enables the acridine-based system to utilize two adjacent cis coordination sites for catalysis. This conformation allows for the elementary step of beta hydride elimination without any ligand dissociation (Fig. 11). As we shall see later, the beta hydride elimination step plays a crucial role in the catalysis observed with these complexes.
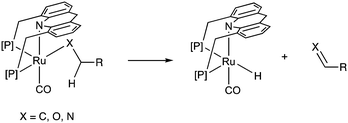 |
| | Fig. 11 Beta hydride elimination in the acridine dearomatized system. | |
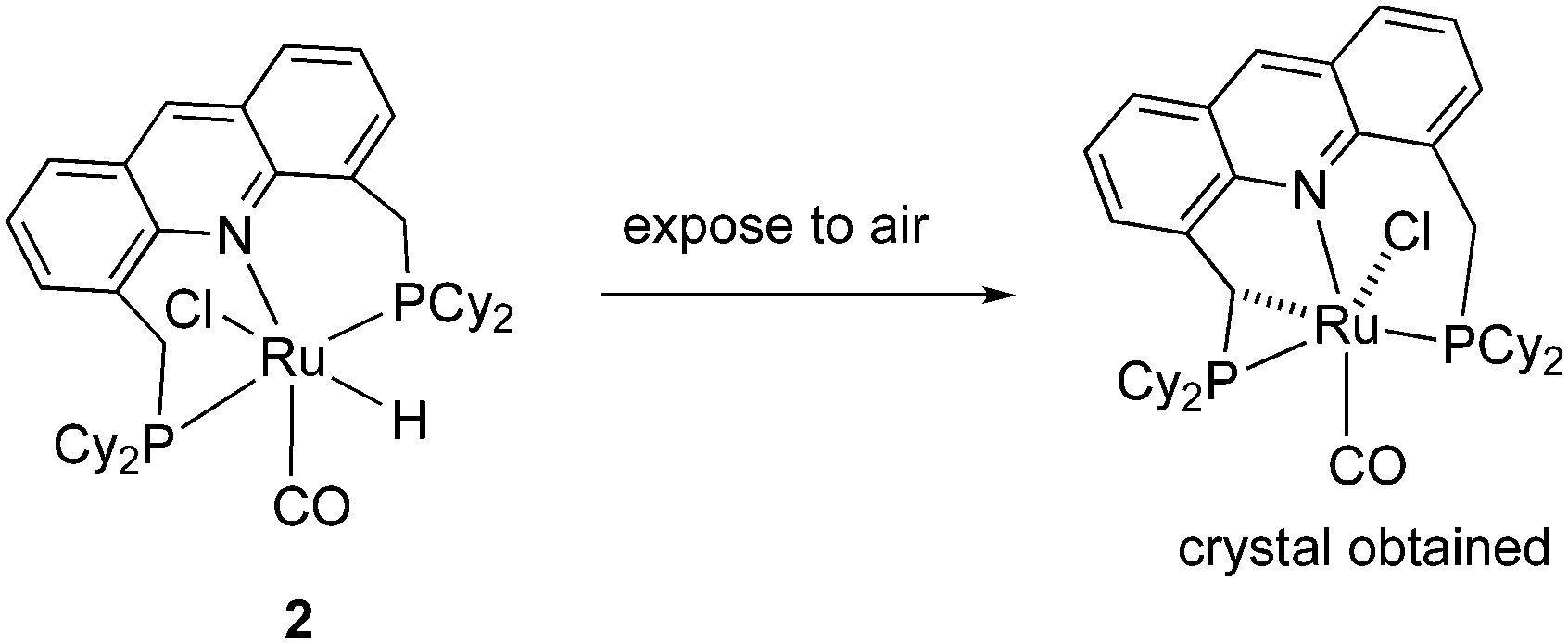
(e) Side arm activation
The activation of the side arm benzylic C–H bond of the acridine ligand by the ruthenium centre is also reported, forming an unusual cyclometalated tetradentate PCNP ligand (Fig. 12).54 However, this feature has not been utilized yet in catalysis using these complexes.
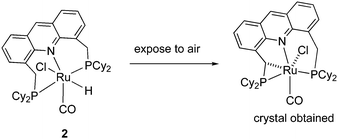 |
| | Fig. 12 Formation of cyclometalated complex. | |
In the subsequent section, we will see how the special reactivities of the acridine system allow it to catalyse unique, sustainable reactions that are not generally obtainable by traditional pincer complexes.
4. Sustainable catalysis with acridine-based pincer complexes
4.1. Reactions related to alcohols and amines
4.1.1. Amination of primary alcohols to primary amines.
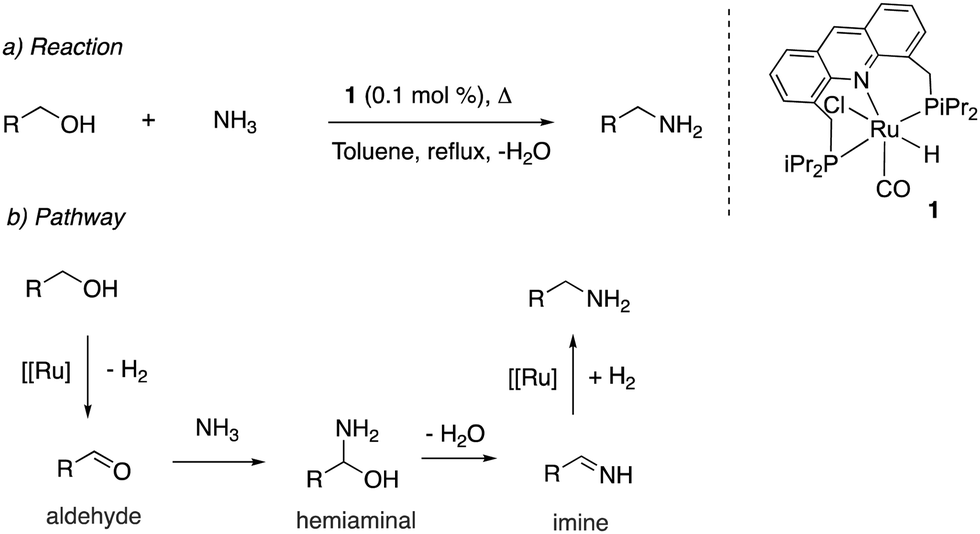
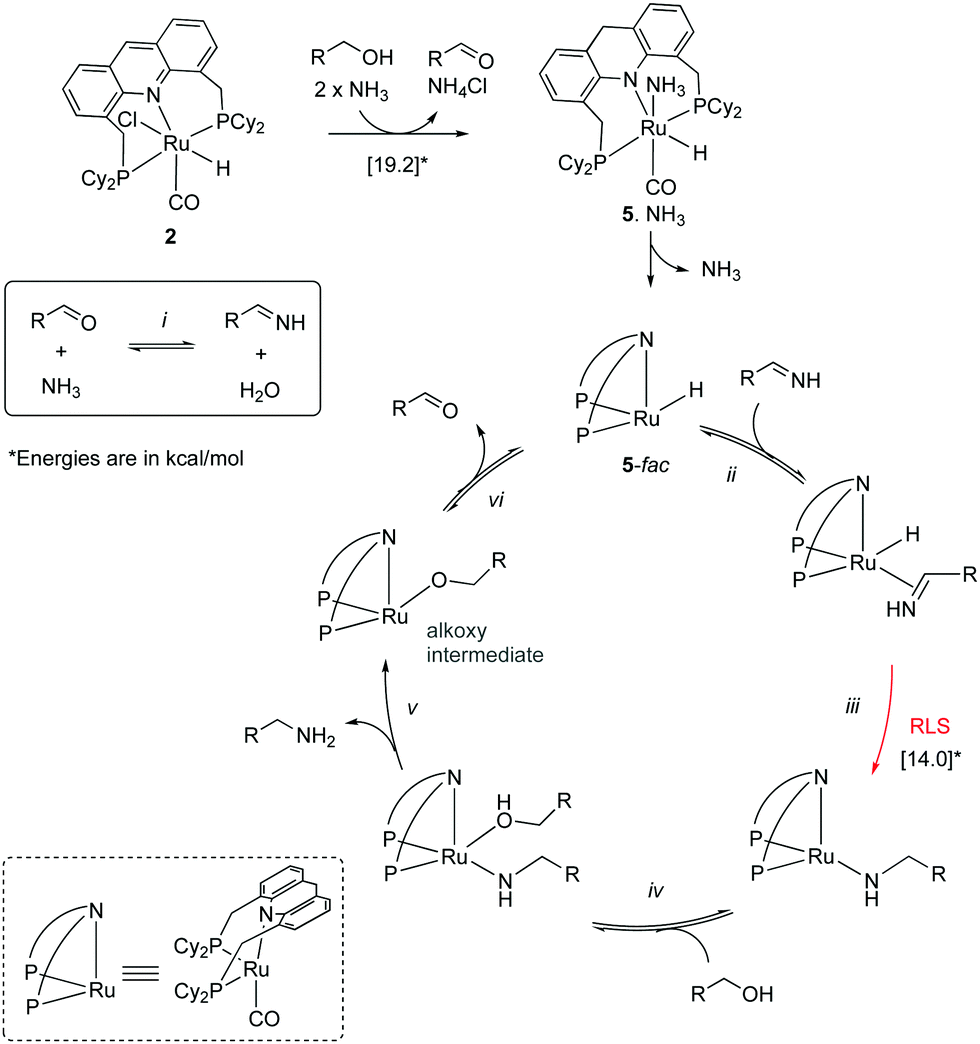
Upon synthesis of the ruthenium acridine complex 1 by our group in 2008, it was employed to synthesize primary amines from primary alcohols and ammonia (Fig. 13a).1 Previous large-scale synthesis of primary amines from alcohols and ammonia utilized heterogeneous catalysts, which required very high temperatures and pressures while generating a mixture of amine products due to the higher nucleophilicity of primary amines than ammonia. Interestingly, the acridine complex 1 catalyzed the conversion at much lower temperatures (135 °C) and NH3 pressure (7.5 bar) while also being highly selective to the formation of primary amines. The water-stable nature of the acridine complex enabled carrying out selected reactions with water as (co)solvent, obtaining the corresponding amine in high to excellent yields. Based on preliminary experimental studies, a hydrogen borrowing reaction pathway was proposed, involving the formation of an aldehyde intermediate, followed by the formation of a hemiaminal intermediate which eliminates water to form an imine intermediate, which reacts with the hydrogen liberated in the first step to form the amine product (Fig. 13b). The reaction of secondary alcohols with ammonia to form secondary amines was later reported with other complexes by different groups with high reactivity and selectivity.61,62
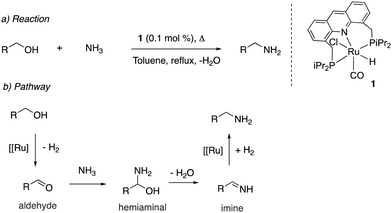 |
| | Fig. 13 Catalytic reaction (a) and proposed reaction pathway (b). | |
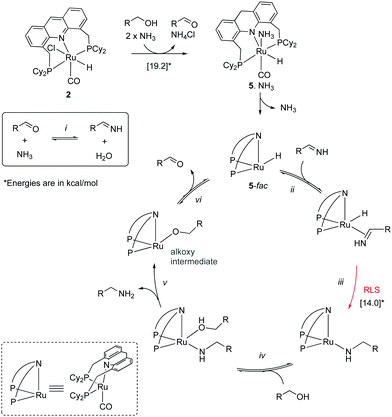 |
| | Fig. 14 Mechanism cycle. | |
4.1.2. Deamination of primary amines to alcohols.
Our group subsequently showed in 2013 that the amination reaction can be reversed, and primary amines can also be converted to primary alcohols and ammonia by heating in dioxane/water in the absence of NH3 (Fig. 15a).63 The deamination of primary amines to alcohols, while facile in biological systems at neutral pH, is challenging synthetically, requiring the use of strong bases such as NaOH. By contrast, using complex 1 as the catalyst, several primary amines were converted to the corresponding alcohols in a neutral dioxane/water reaction medium in moderate to high yields (Fig. 15a). Further, the mechanistic insights obtained during the studies also resulted in the development of a deaminative diamine cyclization procedure to form the corresponding secondary cyclic amines (Fig. 15b).
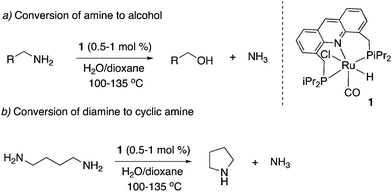 |
| | Fig. 15 Conversion of amines to alcohols (a) and diamines to secondary cyclic amines (b) catalyzed by 1. | |
4.1.3. Selective alcohol dehydrogenation to acetals.
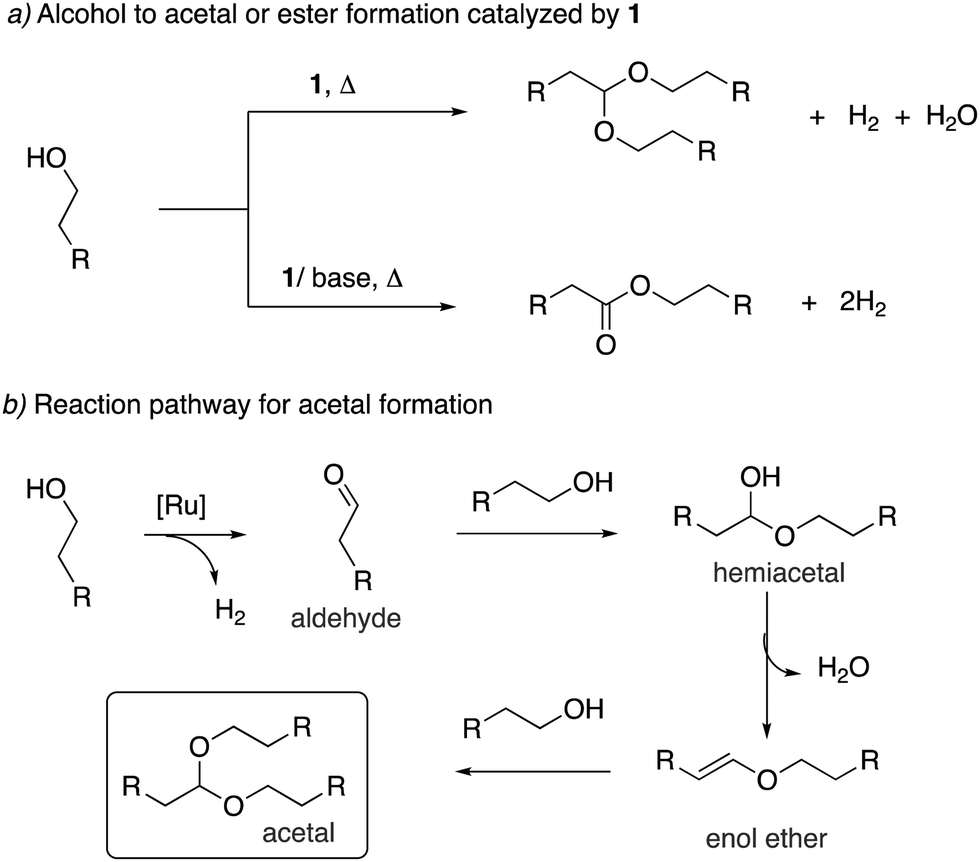
Our group reported in 2009 that the aromatic complex 1 (0.1 mol%) catalyzes the dehydrogenative coupling of primary alcohols to form the corresponding acetals under neutral reaction conditions (Fig. 16a).53 Selective oxidation of alcohols to acetals is challenging as most employed catalysts, including PNN dearomatized pincer complexes, generate esters as the dehydrogenation coupling product instead of acetals.64,65 Interestingly, in the case of complex 1, while the neutral reaction conditions provided the acetal as primary reaction product, addition of one equiv. KOH co-catalyst completely changed the selectivity to ester formation. Experimental observations support a reaction pathway involving the initial generation of an aldehyde intermediate, followed by alcohol addition to generate a hemiacetal intermediate (Fig. 16b). Dehydration of the hemiacetal further generates an enol ether in the acetal formation pathway, whereas the competing hemiacetal dehydrogenation can produce the ester by-product. The formed enol ether generates the desired acetal upon nucleophilic attack of alcohol. This proposed pathway involving an enol ether intermediate was further supported by the fact that in substrates where such enol ether formation was not possible due to the lack of a beta proton, esters were observed as the product. Notably, the pyridine-based aromatic complexes were not active at all under neutral conditions for the reaction, being coordinatively saturated, and the special reactivity of the acridine-based complex was surmised due to the elongated Ru–N bond, possibly imparting a hemilabile quality.
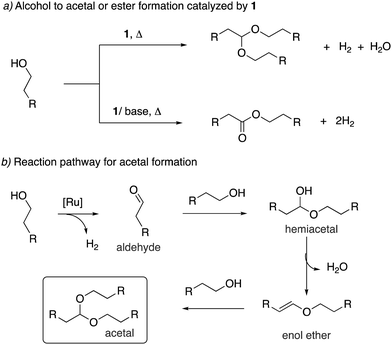 |
| | Fig. 16 (a) Formation of acetal from alcohol catalysed by 1 and (b) the reaction sequence. | |
4.1.4. Formation of N-Heteroaromatics from alcohols and NH3.
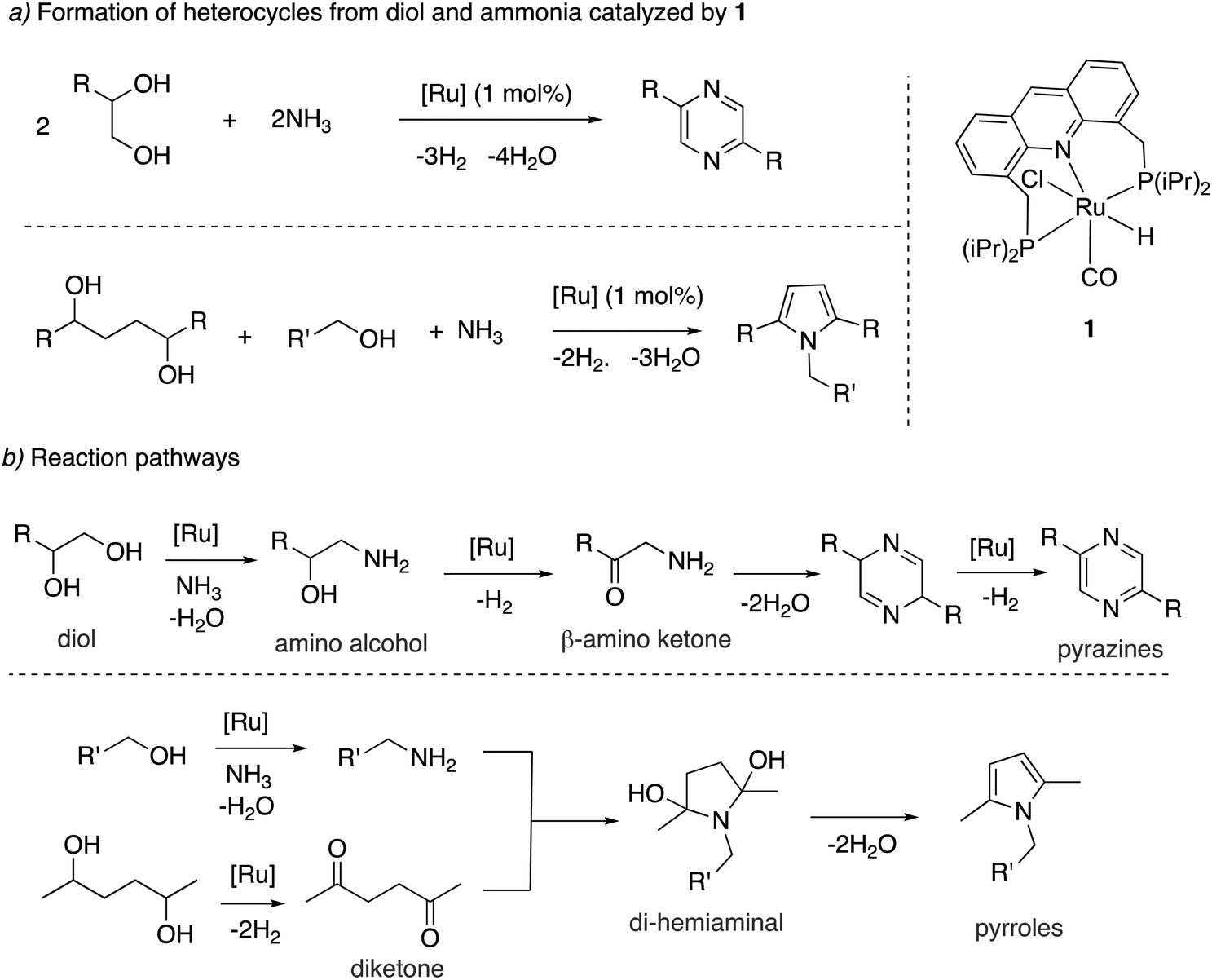
Our group showed in 2018 that the aromatic complex 1 is an effective catalyst for the dehydrogenative direct synthesis of N-heteroaromatics such as pyrroles and pyrazines from alcohols in two/three-component reaction systems using ammonia gas (Fig. 17a).66 Multicomponent systems to access heterocycles with H2 liberation were known previously, but they required the use of amino alcohols or amines as substrates.67–70 The use of ammonia, on the other hand, has a clear economic advantage. Complex 1 (1 mol%) catalyzes the synthesis of pyrazines when 1,2-diol is used as a substrate under 7 bars of ammonia in moderate to high yields. On the other hand, when a 1,4-diol was used, along with another mono-alcohol, the formation of pyrroles was observed. For pyrazine formation from vicinal diols, a mechanism was proposed involving the initial amination of the diol to generate amino alcohol, followed by dehydrogenation to generate beta-amino ketone intermediate (Fig. 17b). Self-condensation of the beta-amino ketone followed by aromatization through Ru-catalyzed dehydrogenation then generates the pyrazine product. In the case of pyrrole synthesis from 1,4-diols and linear alcohols, the linear alcohol is first converted to amine, which then nucleophilically attacks a diketone intermediate (generated from diol via dehydrogenation) to form a di-hemiaminal intermediate. Water liberation from this intermediate subsequently generates the desired substituted pyrrole (Fig. 17b). Control experiments and mechanistic investigation suggested that the employed aromatic catalyst is dearomatized during the reaction to 4, which is the actual catalyst, like with many previously discussed reactions with this system. Notably, the reaction is made possible because complex 1 can catalyze the formation of amines from alcohols and ammonia, which is an essential step of the reaction.
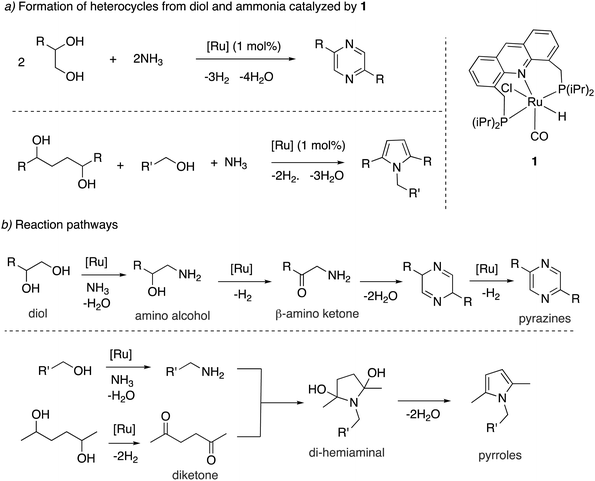 |
| | Fig. 17 (a) Formation of heterocycles from alcohols and ammonia catalyzed by 1, (b) reaction pathways to access pyrazines and pyrroles. | |
4.1.5. Catalytic upgradation of ethanol to higher alcohols.
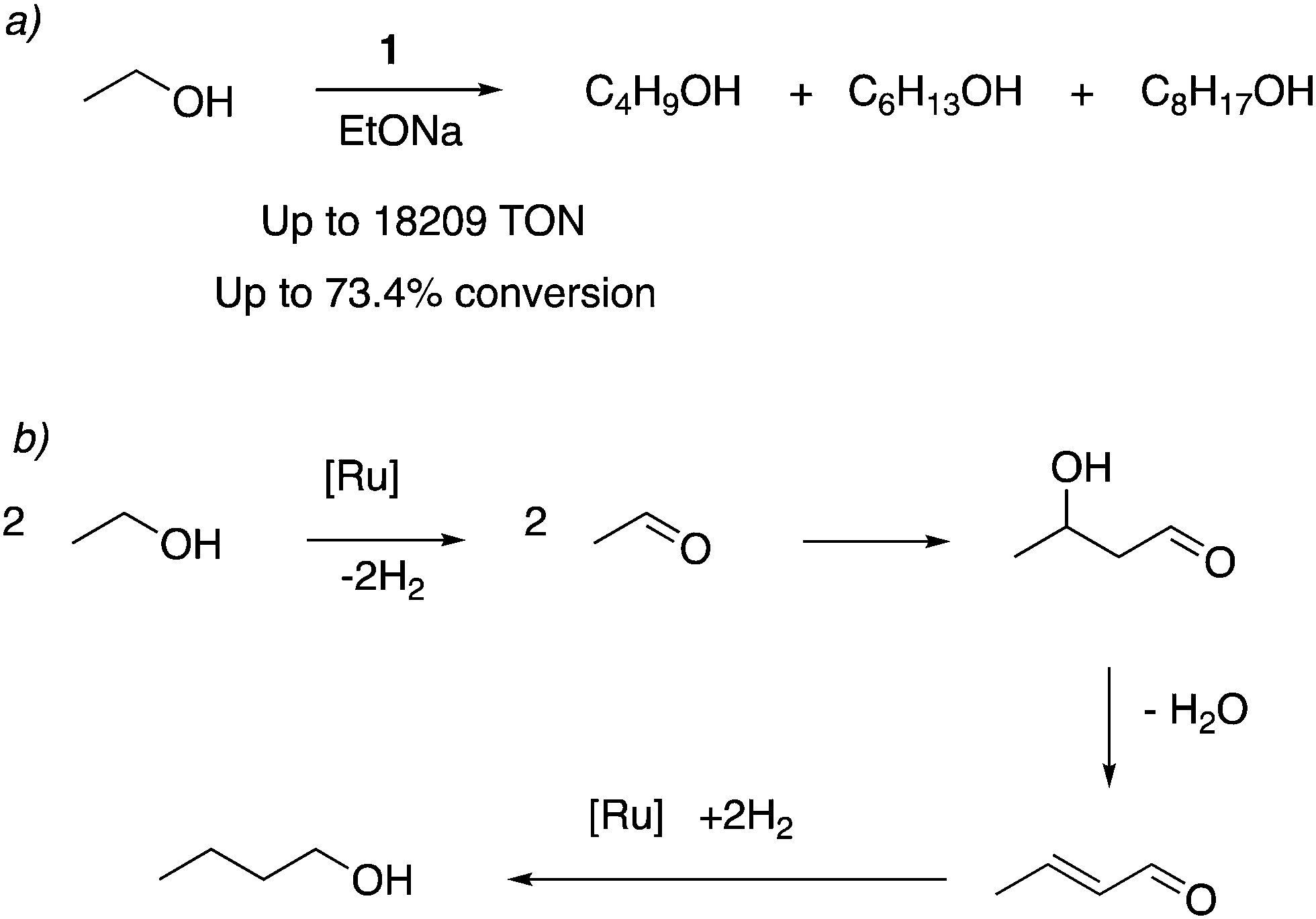
In 2016 our group reported that the dearomatized complex 4 is an excellent catalyst for the upgradation of ethanol to higher biofuels, including butanol, hexanol, and C8 alcohols via the Guerbet process (Fig. 18a and b).71 Ir and Ru based molecular complexes for the Guerbet process for ethanol upgradation were previously reported by other groups, with moderate activity and selectivity.72–75 It was found that even a small amount of 4 (0.001 mol%) can catalyze the ethanol upgradation with high activity (turnover number over 18![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000) and selectivity. Under similar reaction conditions, the acridine-based complexes displayed 4–6 times the catalytic activities than the pyridine-based complexes. The reaction profile showed that in the initial hours, 1-butanol is formed selectively, which can further couple to generate higher molecular weight alcohols as the reaction progresses. Based on prior reports, an aldehyde generation reaction mechanism was proposed that does not involve O–H bond activation via Ru–amido metal–ligand cooperation due to the low basicity of the amido ligand in the dearomatized complex. Instead, a reaction mechanism involving the fac isomer with initial dehydrogenation from coordinated alcohol followed by beta hydride elimination was proposed. To be noted, later reports have also employed manganese-based first row transition metal complexes for ethanol upgradation to butanol.76,77
000) and selectivity. Under similar reaction conditions, the acridine-based complexes displayed 4–6 times the catalytic activities than the pyridine-based complexes. The reaction profile showed that in the initial hours, 1-butanol is formed selectively, which can further couple to generate higher molecular weight alcohols as the reaction progresses. Based on prior reports, an aldehyde generation reaction mechanism was proposed that does not involve O–H bond activation via Ru–amido metal–ligand cooperation due to the low basicity of the amido ligand in the dearomatized complex. Instead, a reaction mechanism involving the fac isomer with initial dehydrogenation from coordinated alcohol followed by beta hydride elimination was proposed. To be noted, later reports have also employed manganese-based first row transition metal complexes for ethanol upgradation to butanol.76,77
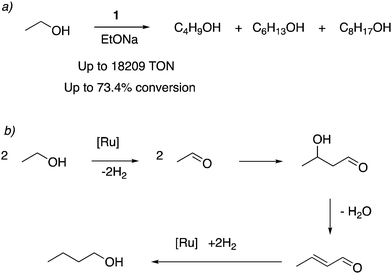 |
| | Fig. 18 (a) Ethanol upgrading to higher alcohols catalyzed by 1. (b) Pathway of Guerbet reaction from ethanol to butanol. | |
4.2. Development of oxidation reactions using water with H2 liberation
4.2.1. Oxidation of cyclic secondary amines to lactams.
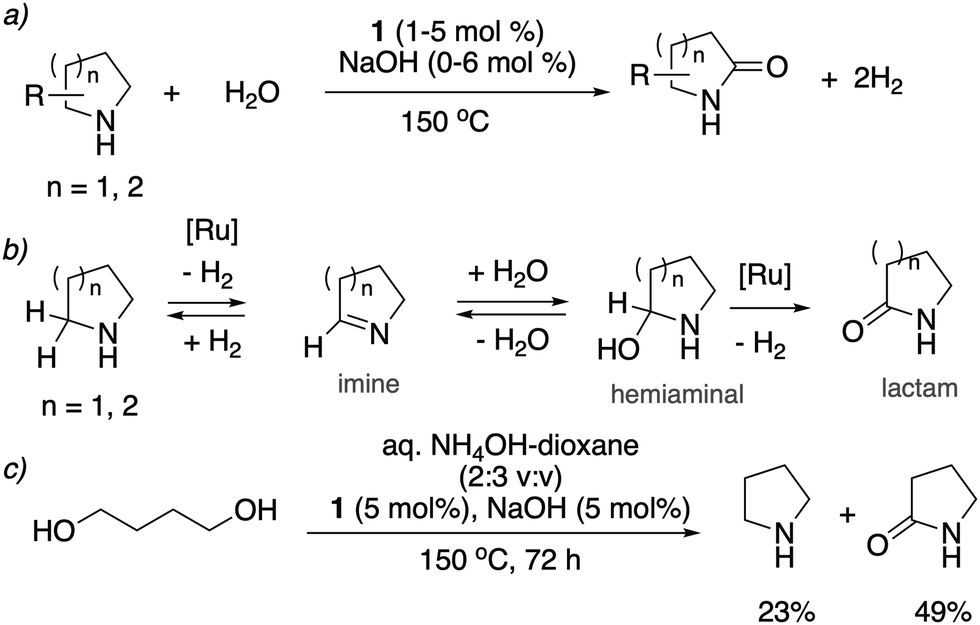
In 2014, our group reported an unprecedented catalytic transformation, namely oxidation of cyclic amines to lactams using water with evolution of two equivalents of H2. Overall, this process results in conversion of the α-methylene group to a C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O group (Fig. 19a).78 The whole process effectively uses water as an oxidant, replacing the previously used strong oxidizers such as iodosobenzene, t-BuOOH and others. The oxidation is catalyzed by the aromatic complex 1 as pre-catalyst in the presence of NaOH co-catalyst. Using this method, the oxidation of several substituted pyrrolidines, piperidines, pyrazine and morpholine to their corresponding lactams with moderate to high yields was demonstrated. Based on control experiments, a reaction pathway involving the initial dehydrogenation of cyclic amines to imines, followed by water addition to generate a hemiaminal with subsequent dehydrogenation to the desired lactam was proposed (Fig. 19b). The proposed mechanism was further verified by H/D exchange experiments ensuring initial imine formation and using 18O labelled water as the solvent, which clearly showed the incorporation of an 18O atom into the product lactam. Finally, combining the previous report of cyclic amine formation from diols, with the newly developed oxidation of cyclic amines to lactams by water, enabled the direct formation of lactams from linear diols and ammonia via the in situ generated cyclic secondary amine (Fig. 19c).
O group (Fig. 19a).78 The whole process effectively uses water as an oxidant, replacing the previously used strong oxidizers such as iodosobenzene, t-BuOOH and others. The oxidation is catalyzed by the aromatic complex 1 as pre-catalyst in the presence of NaOH co-catalyst. Using this method, the oxidation of several substituted pyrrolidines, piperidines, pyrazine and morpholine to their corresponding lactams with moderate to high yields was demonstrated. Based on control experiments, a reaction pathway involving the initial dehydrogenation of cyclic amines to imines, followed by water addition to generate a hemiaminal with subsequent dehydrogenation to the desired lactam was proposed (Fig. 19b). The proposed mechanism was further verified by H/D exchange experiments ensuring initial imine formation and using 18O labelled water as the solvent, which clearly showed the incorporation of an 18O atom into the product lactam. Finally, combining the previous report of cyclic amine formation from diols, with the newly developed oxidation of cyclic amines to lactams by water, enabled the direct formation of lactams from linear diols and ammonia via the in situ generated cyclic secondary amine (Fig. 19c).
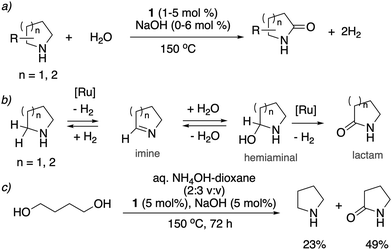 |
| | Fig. 19 (a) Cyclic amines to lactams using water as oxidant catalyzed by 1, (b) reaction pathway (c) accessing lactams directly from diols and ammonia. | |
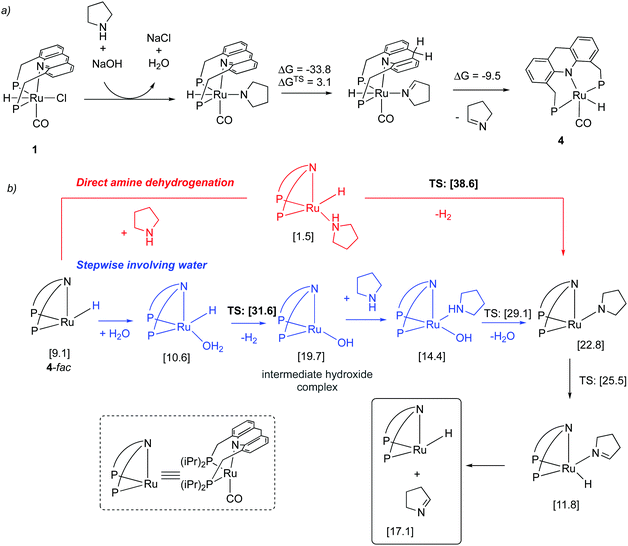 |
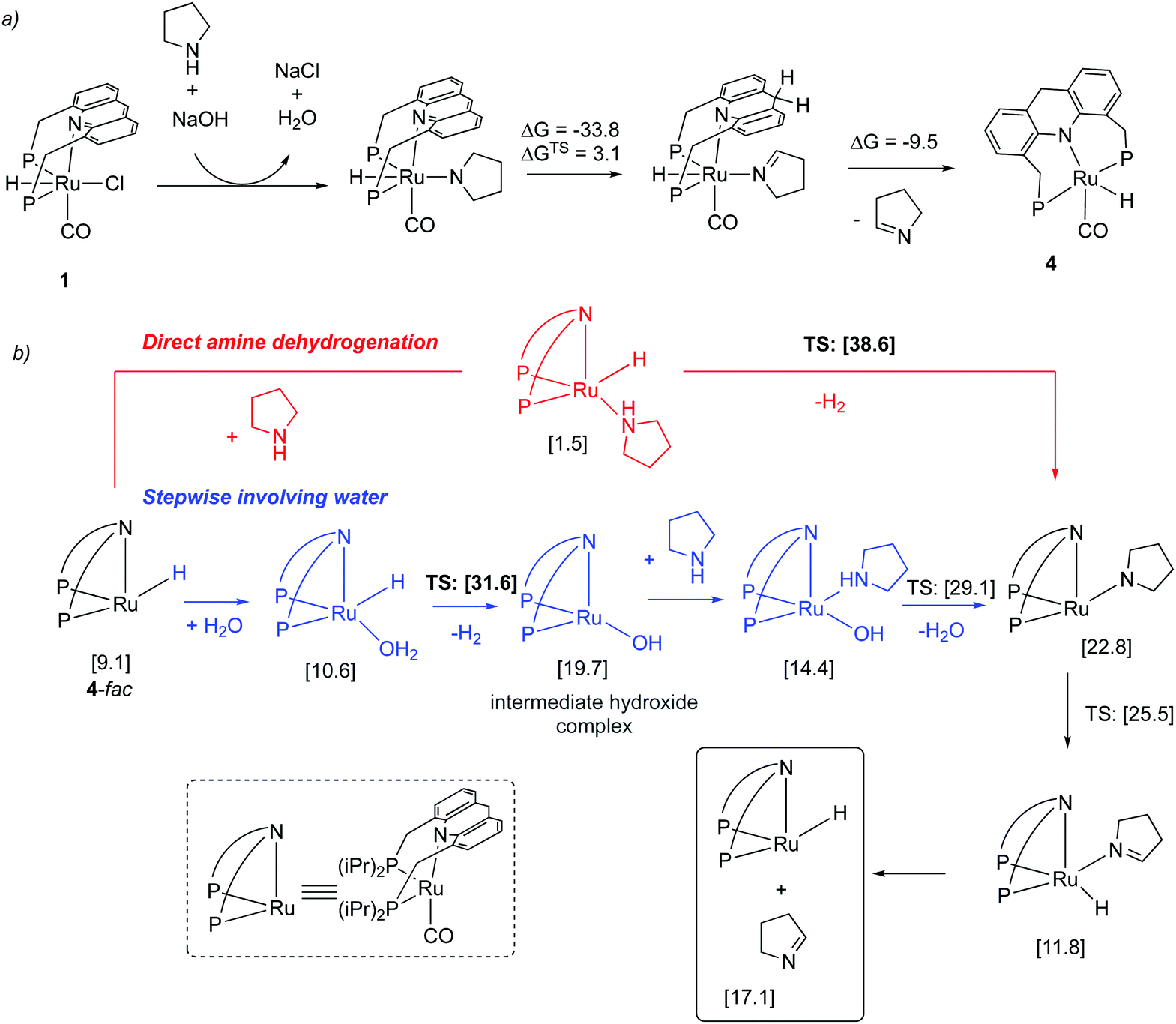
| | Fig. 20 (a) Initial dearomatization of complex 1 (b) amine dehydrogenation via direct (red) and stepwise pathway involving water (blue). Calculated free energies (in brackets) are in kcal mol−1 with respect to 4-mer + amine + water. | |
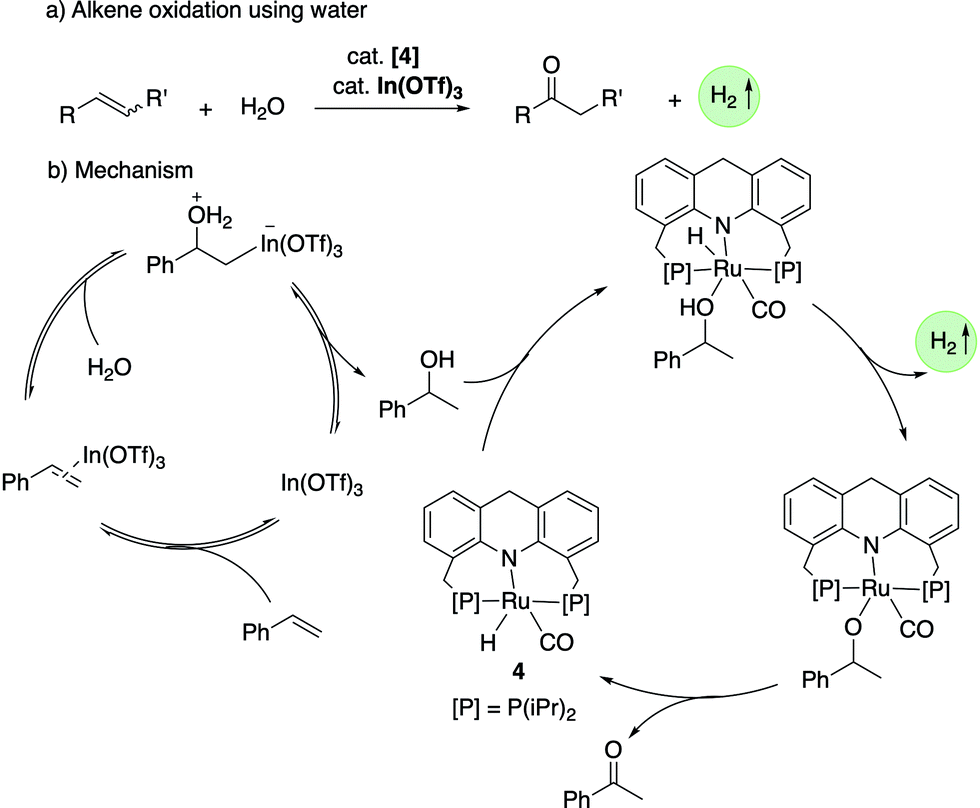
4.2.2. Markovnikov oxidation of alkenes to ketones using water.
In 2020, our group demonstrated the Markovnikov oxidation of alkenes by water to the corresponding ketones using the acridine-based complexes as catalysts along with In(OTf)3 co-catalyst (Fig. 21a).79 As opposed to the well-established Wacker oxidation process to obtain ketones from alkenes, which requires O2 as terminal oxidant, the newly developed process does not require a terminal oxidant and is associated with the liberation of one equiv. H2. The In(OTf)3 co-catalyst (1–40 mol%) is necessary for the initial hydration of alkenes to secondary alcohols (Fig. 21b). The generated alcohol is dehydrogenated in situ by the ruthenium complex (1.5 mol%) to form the corresponding ketone. Isotopic labelling studies verified that the H2O oxygen atom is incorporated into the ketone product. Styrenes, 2-vinylnaphthalene and 4-vinylbiphenyl produced the corresponding ketones in moderate to high yields. Other than styrenes, strained cyclic alkenes such as norbornene, cycloheptene and cyclooctene were also amenable to the transformation.
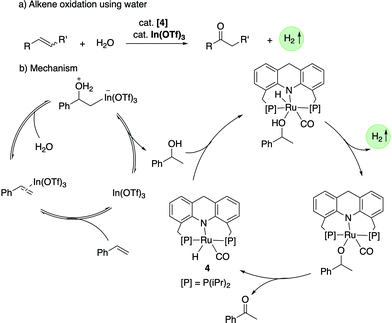 |
| | Fig. 21 (a) Alkene oxidation to a ketone with water catalyzed by 4. (b) Reaction mechanism. | |
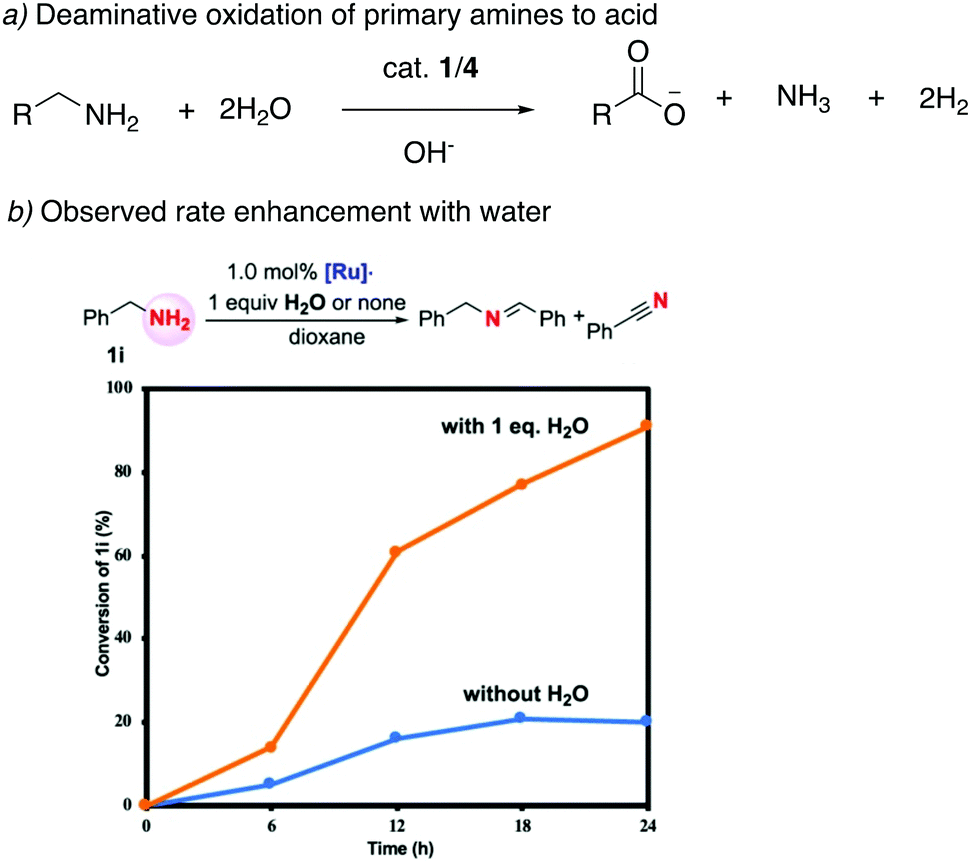
4.2.3. Deaminative oxidation of amines to acids.
Our group employed the acridine-based complexes to achieve a unique oxidative deamination process for amines using alkaline water, which generate carboxylate salts, H2, and ammonia as the reaction products (Fig. 22a).56 Oxidative deamination is a well-known process in biological systems, catalyzed by enzymes such as amine dehydrogenases or copper amine oxidases. On the other hand, carrying out the same transformation synthetically is challenging and prior to this report required the use of strong stoichiometric oxidants. Based on control experiments, a reaction pathway was proposed involving the initial formation of imine through amine dehydrogenation, followed by deaminative imine hydration to aldehyde, with subsequent aldehyde oxidation to acid being the final step. Interestingly, the amine dehydrogenation rate, catalyzed by the dearomatized complex 4, increased more than fourfold when water was present in the system (Fig. 22b). DFT studies revealed that a water-assisted amine dehydrogenation pathway takes place in the presence of water, involving formation of a Ru–hydroxy complex, which is more favourable than direct amine dehydrogenation. Further, while primary alcohols yield carboxylate salts under the reaction conditions, secondary amines produce ketones as the deaminative oxidative product. Moreover, amides also show reactivity to generate carboxylic acid products.
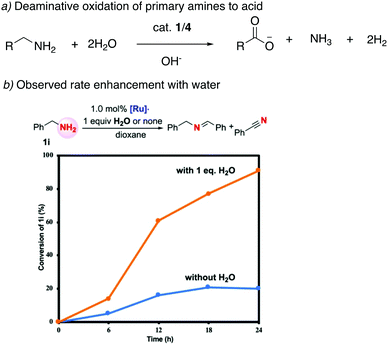 |
| | Fig. 22 (a) Deaminative oxidation of primary amines catalyzed by 1/4. (b) rate enhancement observed in the presence of water. Fig. 22b is adapted with permission from ref. 56. Copyright 2020 American Chemical Society. | |
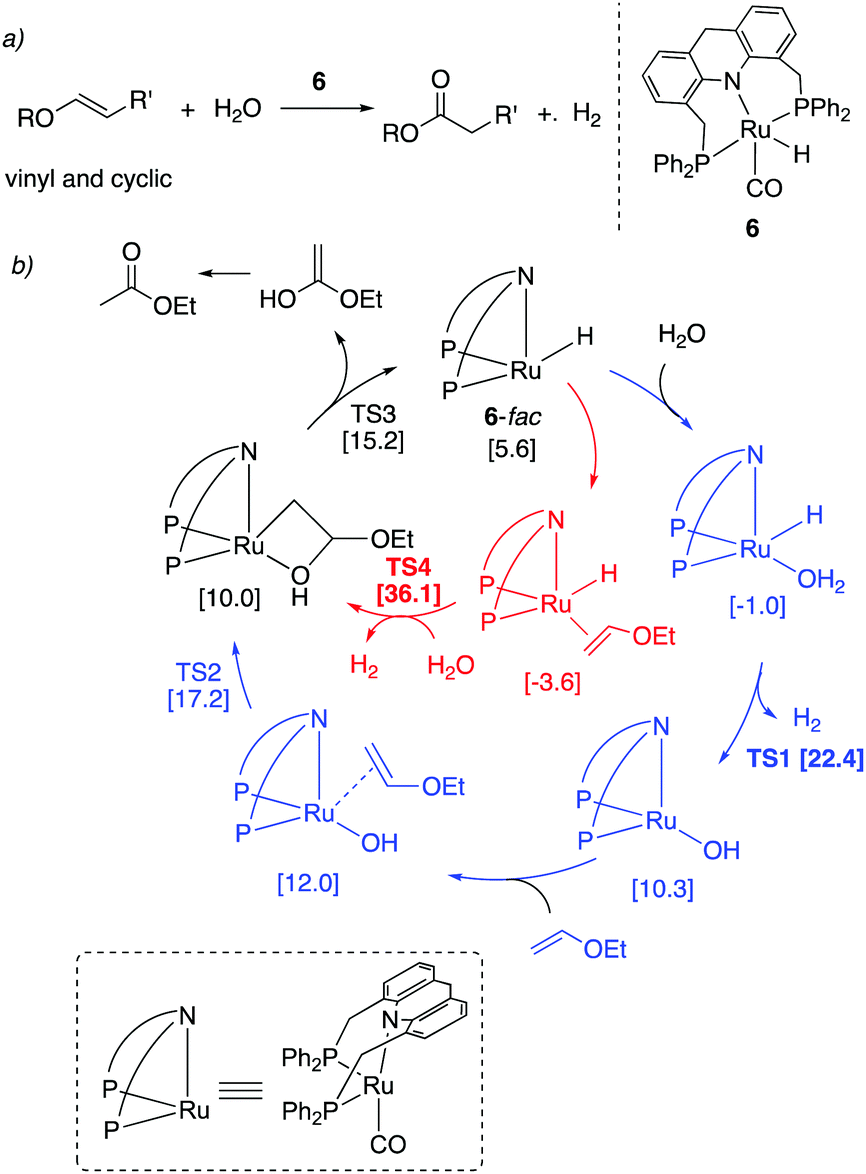
4.2.4. Oxidation of enol ethers to esters.
We have recently shown that the Ru–acridine-based PNP pincer complexes also catalyze the oxidation of vinyl and cyclic enol ethers to their corresponding esters using water as the oxidant while generating H2 gas as the reaction by-product (Fig. 23a).59 Interestingly, the dearomatized acridine complex catalyzed both the hydration and dehydrogenation steps of the reaction without the presence of any further additives. While the acridine(iPr) complex 4 produced noticeable amounts of hydrogenated side-products, the oxidation was selective when the analogous acridine(Ph) (6) was used as catalyst (1–1.5 mol%), producing esters in good to excellent yields. The oxidation of vinyl ethers proceeded at 125 °C, whereas the oxidation of cyclic enol ethers required a higher temperature (150 °C). The oxidation of linear internal enol ethers did not proceed due to their lower coordinating nature. Interestingly, DFT calculations suggested an inner-sphere coupled stepwise hydration dehydrogenation reaction pathway (in blue) as opposed to an outer-sphere hydration dehydrogenation mechanism (in red, Fig. 23b). The reaction did not proceed when traditional PNN-based rigid Ru–pincer complexes were used due to the formation of practically irreversible hydroxy complexes.
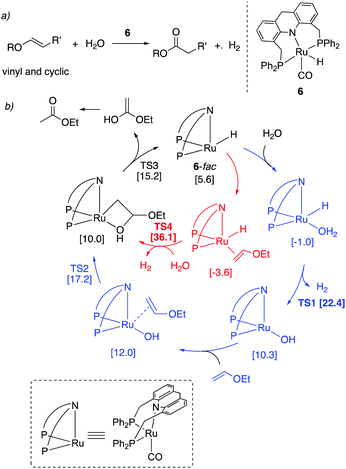 |
| | Fig. 23 (a) Enol ether to ester conversion catalyzed by complex 6. (b) Reaction mechanism with stepwise inner-sphere and direct outer-sphere pathways. Calculated free energies (in brackets) are in kcal mol−1 with respect to 6-mer + ethyl vinyl ether + water. | |
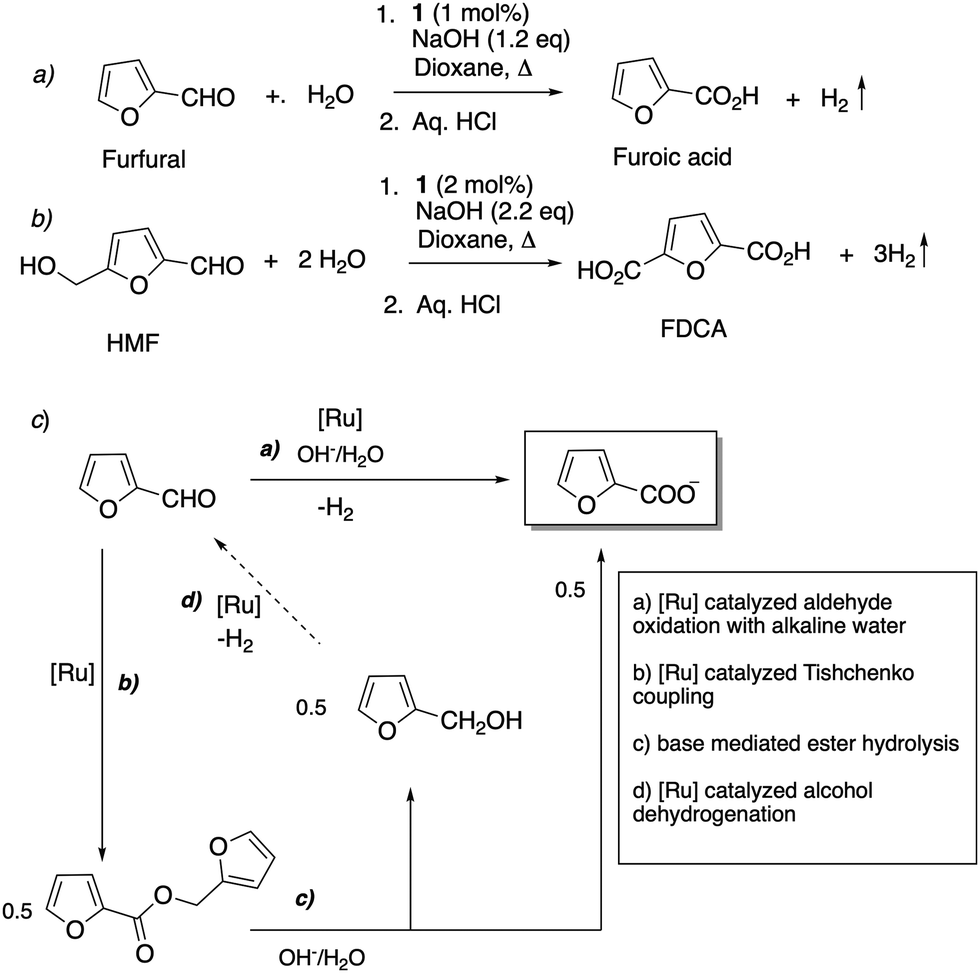
4.2.5. Oxidation of biomass derived furfural and 5-hydroxymethyl furfural.
Very recently, in 2022, we reported that the acridine based Ru–PNP complexes 1 and 2 are excellent catalysts for the oxidation of furfural and 5-hydroxymethyl furfural (HMF) to furoic acid and 2,5-furandicarboxylic acid (FDCA), respectively, using alkaline water as the oxidant (Fig. 24a and b).80 Furfural and HMF are bio-based renewable feedstocks, and their oxidation products are important for the implementation of future green chemical industry.81,82 It was found that while in the presence of other pyridine-based pincer complexes furfural and HMF degraded to unidentifiable polymers, the acridine-based complexes selectively oxidized them with high yields (>95%), while suppressing decomposition. Mechanistic experiments indicated that the employed aromatized complex is dearomatized in the initial step to generate complex 4, which subsequently catalyses the reaction. The background decomposition suppression was assigned to the ability of complex 4 to disproportionate the reactant aldehyde to acid and alcohol via a sequence of Ru-catalysed Tishchenko coupling followed by base mediated ester hydrolysis, quickly consuming all the aldehyde before decomposition can take place (Fig. 24c). Using complex 1 and alkaline water, furfural/HMF were oxidized to furoic acid/FDCA and H2 with >95% yield with the scalability of the process demonstrated by a gram-scale reaction.
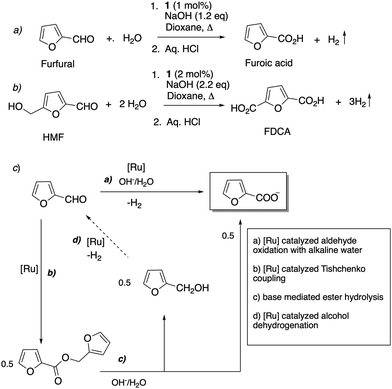 |
| | Fig. 24 (a) Furfural oxidation to furoic acid with water (b) HMF oxidation to FDCA using water (c) reaction pathway including initial Tishchenko coupling of furfural. | |
4.3. Development of liquid organic H2 carrier systems
4.3.1. Ethylene glycol as a H2 carrier.
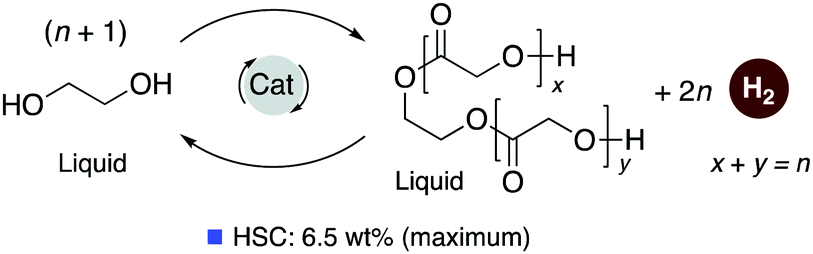
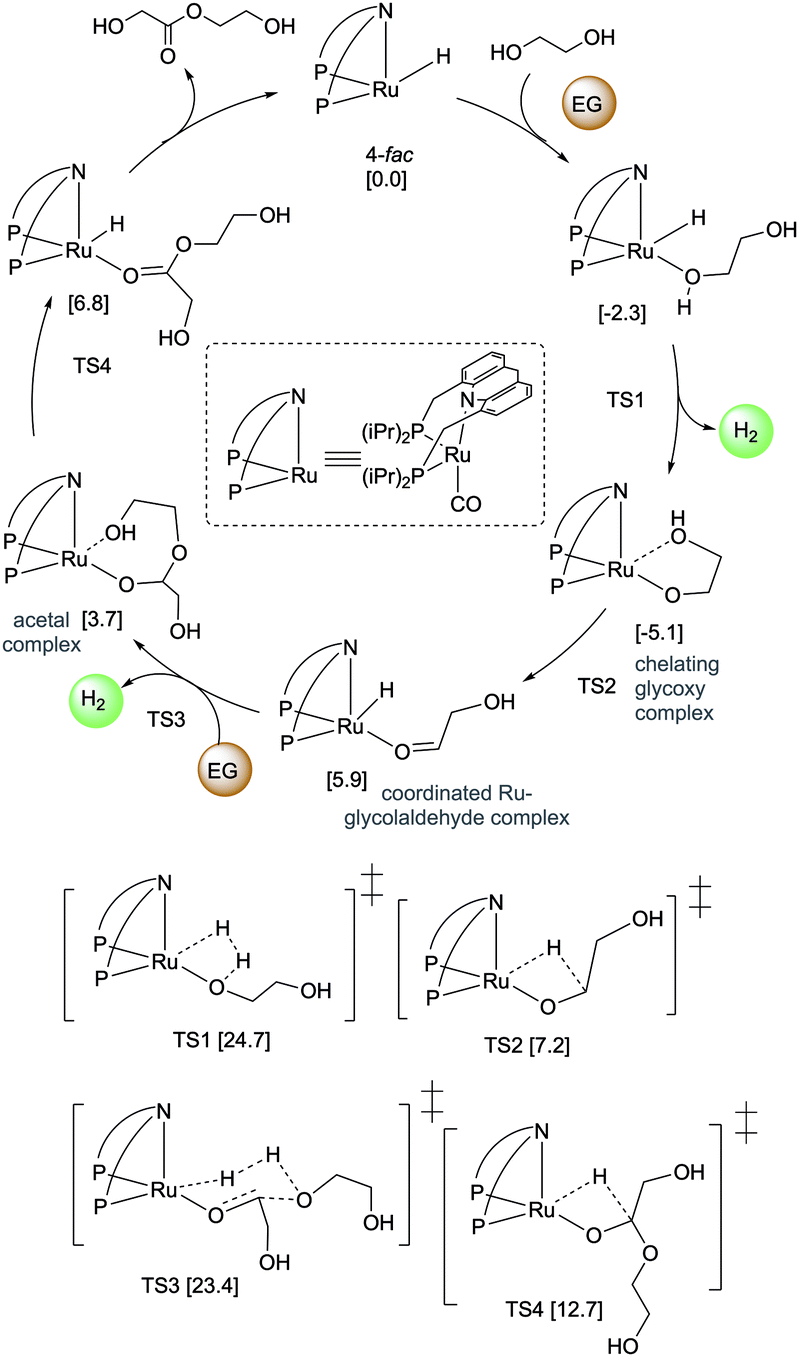
Pincer-based catalysts have been used in recent times to develop amine and alcohol-based liquid organic hydrogen career systems. In 2019, our group reported a reversible LOHC system based on ethylene glycol (EG) using the ruthenium acridine complexes as catalysts with 1 mol% catalyst loading (Fig. 25).83 Ethylene glycol is a widely produced chemical derived from fossil fuels and biomass and is commonly used in antifreeze formulation, polymer synthesis, and others. Complex 4 catalyzes the dehydrogenative self-coupling of ethylene glycol to generate hydrogen up to a theoretical maximum of 6.5 wt%. The resulting H2 lean products consist of various non-volatile liquid oligoesters. The same catalyst (1 mol% loading) can convert the oligoesters to EG by hydrogenation under H2 pressure (40 bar). The acridine-based dearomatized complex 4 showed the highest activity among the screened pincer complexes, although other pyridine-based complexes were also active to different extents. DFT calculations elucidated the plausible mechanism of the process (Fig. 26) indicating that the fac isomers are likely involved in the active catalytic cycle. In the presence of 4, H2 liberation from the Ru-coordinated ethylene glycol initially generates a chelating glycoxy complex, followed by beta hydride elimination to form a coordinated Ru–glycolaldehyde complex. Nucleophilic attack of another EG molecule on the bound aldehyde has a low energy barrier (23.4 kcal mol−1 with respect to the fac isomer of 4). Interestingly, the corresponding transition state (TS3) is a six-membered Zimmerman–Traxler-like transition state and is also associated with simultaneous H2 liberation. Further beta hydride elimination from the resulting acetal complex generates the dimerized ester product, which can further couple to produce higher oligomers.
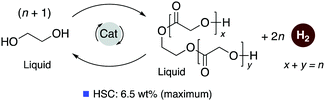 |
| | Fig. 25 Liquid organic hydrogen carrier system based on EG. | |
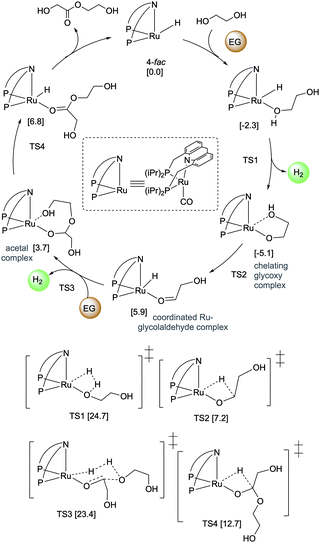 |
| | Fig. 26 Mechanism of EG dehydrogenative coupling. Calculated free energies are in kcal mol−1 with respect to 4-fac + 2*EG. | |
4.3.2. Reversible H2 carrier based on ethylene glycol and ethanol.
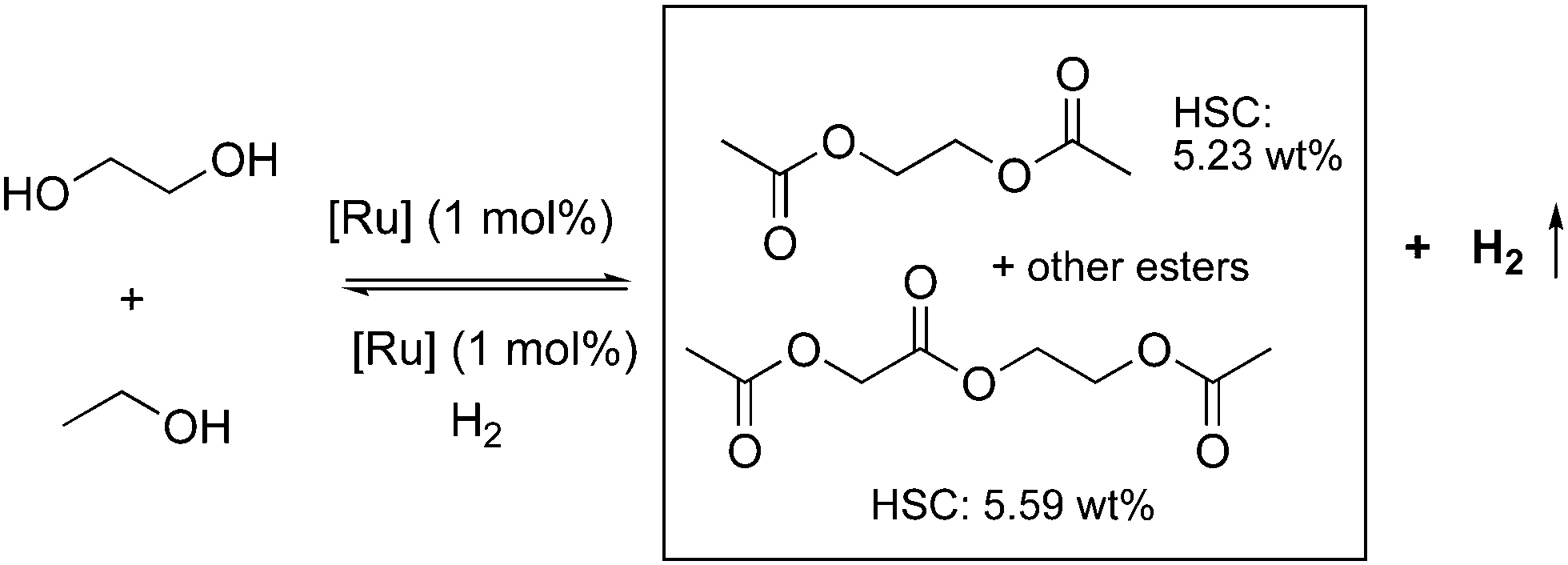
In the subsequent year, our group reported another LOHC system using the acridine-based complexes employing two widely available inexpensive alcohols— ethylene glycol and ethanol (Fig. 27).84 The EG and ethanol undergo dehydrogenative coupling at 150 °C in the presence of 4 (1 mol%), generating hydrogen with high yields (87%), along with an H2 lean liquid esters mixture. The main components of the liquid esters were ethylene glycol diacetate and ethyl acetate, along with some ethylene glycol monoacetate 2-acetoxyethyl-2-acetoxyacetate, and others. The dehydrogenation also proceeds under neat conditions without any solvent and can theoretically achieve ∼5.5 wt% of H2 storage density. Interestingly, the H2 lean esters can be reloaded with H2 under hydrogen pressure by the same catalyst (1 mol%, 50 bars) or by other efficient hydrogenation catalysts with an H2 pressure as low as 5 bars, making the whole process inexpensive and operable under mild conditions.
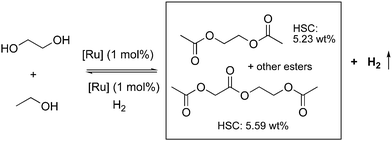 |
| | Fig. 27 Liquid organic hydrogen carrier system based on EG and ethanol. | |
4.3.3. Neat formic acid dehydrogenation.
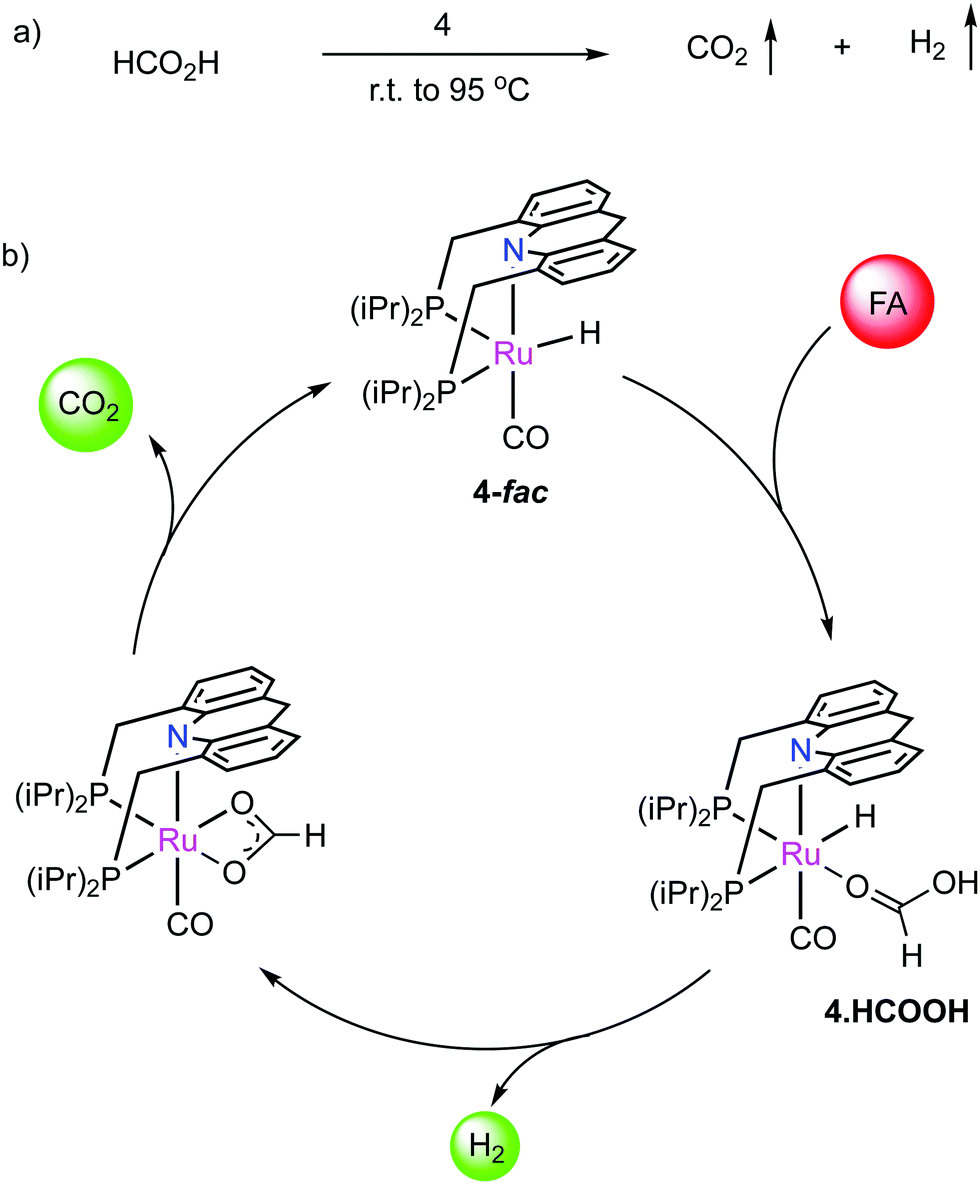
We have reported that the dearomatized acridine complex 4 is highly effective for the additive-free dehydrogenation of neat formic acid (FA), reaching a turnover number (TON) over 1.7 million (Fig. 28a).85 Formic acid is a promising H2 carrier for its use in the transportation sector, however, most H2 liberation systems from formic acid require solvents and/or additives,86,87 which decreases the practical applicability, with only handful reports for neat FA dehydrogenation.88–90 Complex 4 showed excellent activity in additive-free neat FA dehydrogenation achieving turnover frequencies (TOFs) over 3000 h−1. The catalyst is also effective in producing H2 from commercially available low-grade 85% FA, and at the same time, can also generate high-pressure H2/CO2 from neat FA (100 bars). The CO contamination in the generated gas flow is minimal (<20 ppm), and the overall system is highly scalable (demonstrated with a kilogram scale experiment). Based on mechanistic investigations, we proposed a reaction sequence involving initial H2 liberation from formic acid by 4-fac, followed by CO2 liberation via beta hydride elimination (Fig. 28b). Computational studies revealed that the beta hydride elimination is the rate-determining step, as verified by kinetic isotope effect studies. The remarkable simplicity of the system, which contains only complex 4 and FA as reactants, makes the system promising for scale-up.
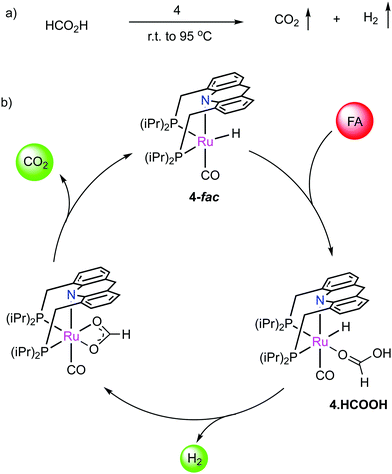 |
| | Fig. 28 (a) Neat formic acid dehydrogenation catalyzed by 4, and (b) reaction mechanism. | |
4.3.4. Methanol reforming.
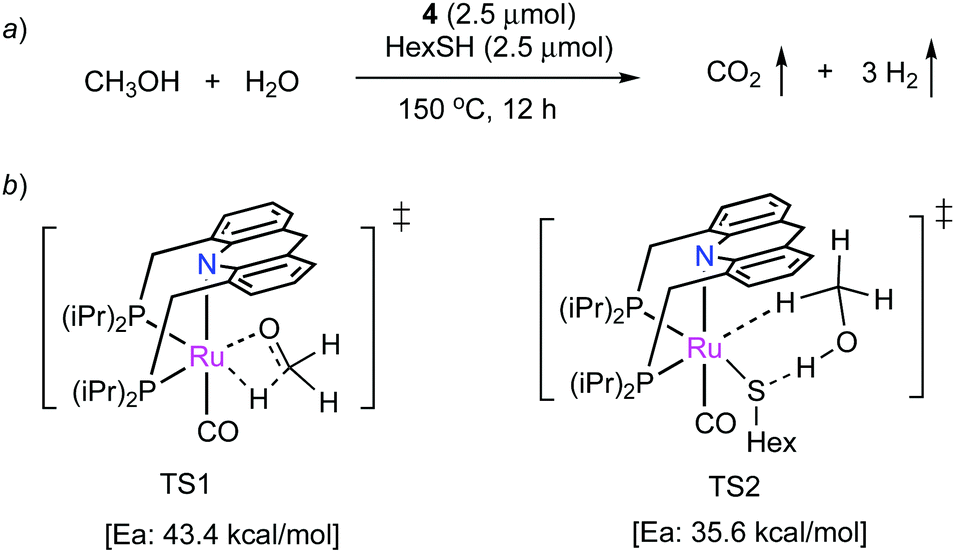
Very recently, our group has demonstrated that the dearomatized complex 4 can also produce H2 from a neat methanol–water solution in the presence of catalytic thiol additive (Fig. 29a).91 H2 generation from methanol reforming is important as it can provide high gravimetric H2 content (>12 wt%). Interestingly, complex 4 without the thiol additive exhibited low activity in the dehydrogenation, which was attributed to the difficulty in initiating methanol dehydrogenation by complex 4 itself, which based on DFT studies involves an inner-sphere transition state (TS1, Fig. 29b) with activation energy of 43.4 kcal mol−1. Quite remarkably, in the presence of catalytic thiol, a thiolate complex is generated, which is much more active for methanol dehydrogenation, allowing the reaction to proceed via a facile outer-sphere mechanism with the assistance of thiolate ligand (TS2, Ea: 35.6 kcal mol−1), increasing the overall activity by nearly two orders of magnitude. The resulting formaldehyde then generates formic acid in the presence of water with H2 liberation, followed by formic acid dehydrogenation to CO2. A hydrogen production TON of 130000 was obtained (highest for aqueous methanol reforming under base-free conditions92–94), with TOFs up to 630 h−1, by heating a solution of methanol, water, and catalytic 4 with catalytic hexanethiol for several weeks, showing the robustness of the system. Notably, the thiol assisted dehydrogenation rate increase is a unique feature of the acridine system due to its ability to adopt a fac structure and is not replicable in other traditional pincer systems such as bipyridyl Ru–PNN complex.
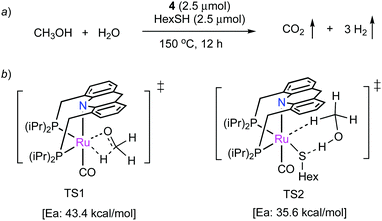 |
| | Fig. 29 (a) Methanol reforming catalyzed by 4, (b) Methanol dehydrogenation—inner-sphere (TS1) and outer-sphere (TS2) with the assistance of thiolate ligand. | |
4.4. Reactions with alcohols and thiols
4.4.1. Thioester synthesis from alcohols and thiols.
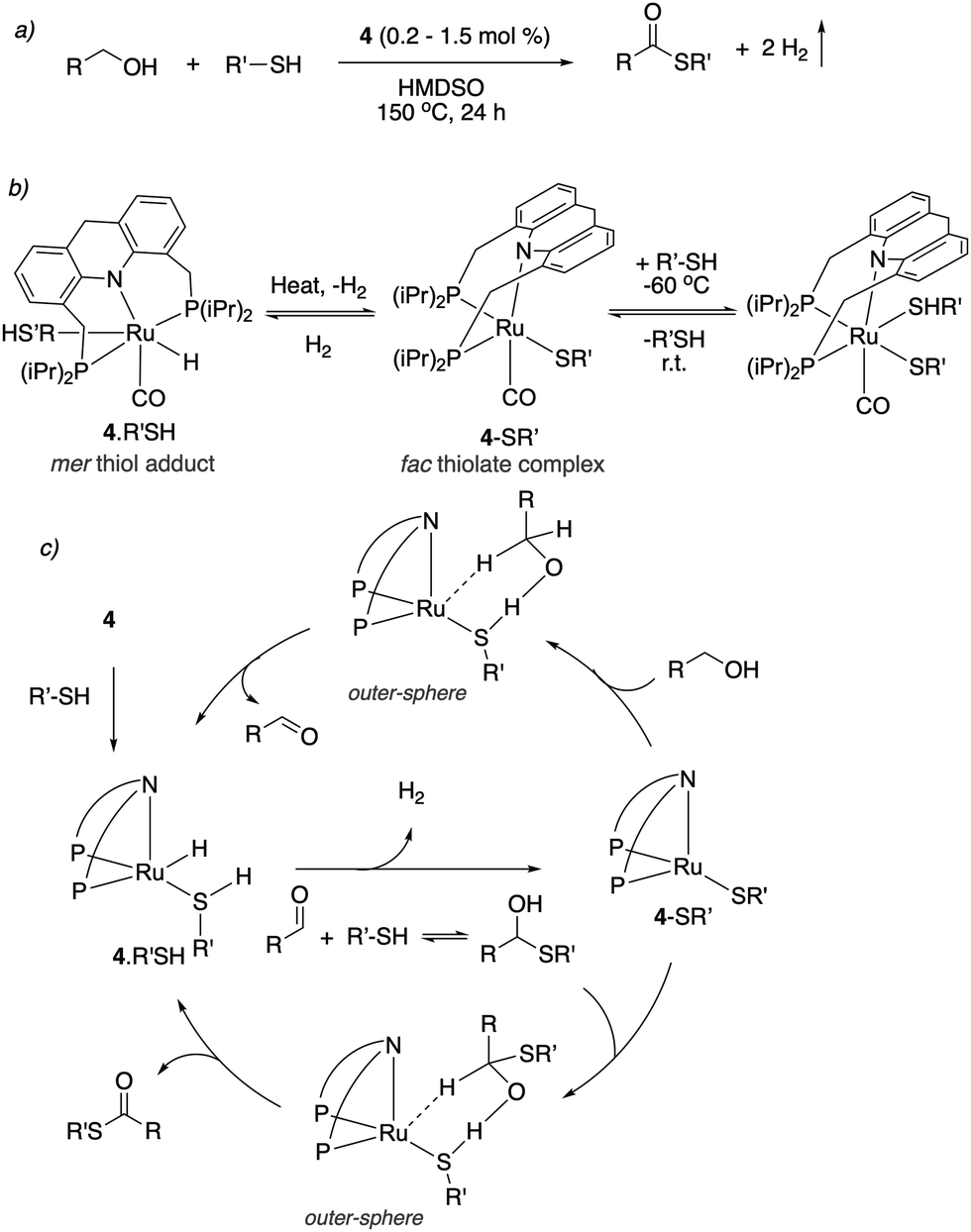
In 2020, our group demonstrated the direct formation of thioesters by the dehydrogenative coupling of alcohols and thiols using the dearomatized ruthenium acridine complex 4 as catalyst (Fig. 30a).58 Thioesters are valuable building blocks and intermediates in formation of heteroaromatics and many other advanced materials, however, their synthesis mainly relied on the acylation of thiols. Direct dehydrogenative coupling of alcohols and thiols has previously been challenging as the traditional pincer complexes are deactivated in the presence of thiol due to its enhanced acidity. On the contrary, complex 4 showed unique reactivity with thiols. At low temperature, the thiol adds to 4 to form the mer-adduct complex (Fig. 30b). When warmed up to room temperature, H2 can be liberated from the complex via a fac isomer, generating the fac thiolate complex, which was observed by NMR (Fig. 30b). It was observed that the thiolate complex can uniquely catalyze an outer-sphere alcohol dehydrogenation with the assistance of a thiolate ligand (Fig. 30c). The resulting aldehyde forms the hemithioacetal in the presence of nucleophilic thiol. Subsequently, outer sphere dehydrogenation of the hemithioacetal by the thiolate complex generates the desired product thioester. Using complex 4, several alcohols and thiols were converted to the corresponding thioesters. Interestingly, the reaction is also operational when an aldehyde, instead of an alcohol, was used as a substrate.
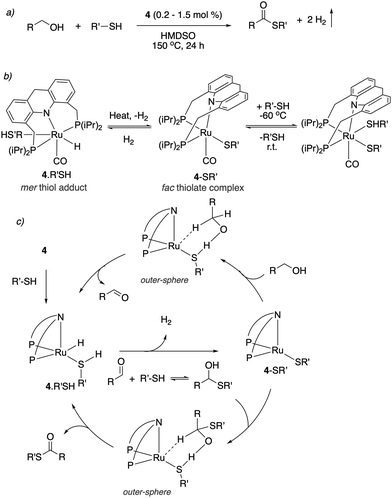 |
| | Fig. 30 (a) Thioester formation from acceptorless dehydrogenative coupling of alcohols and thiols (b) reactivity of complex 4 with thiol. (c) Proposed reaction mechanism. | |
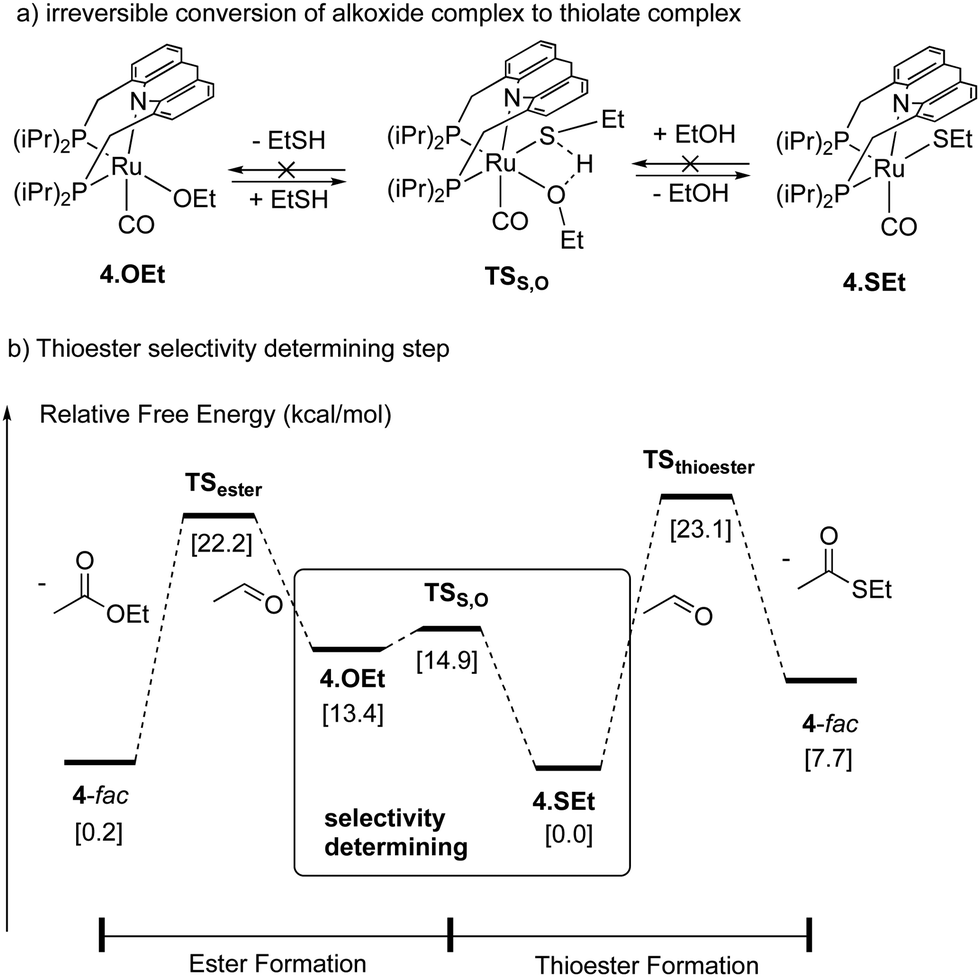
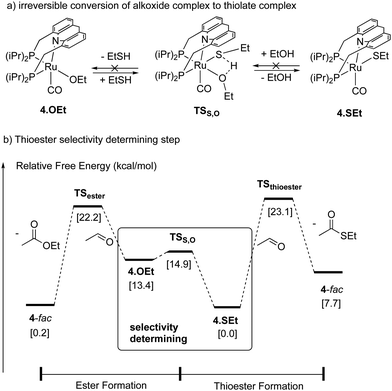 |
| | Fig. 31 (a) Conversion of ethoxide complex to thiolate complex (b) selectivity determining step in the selective thioester formation. | |
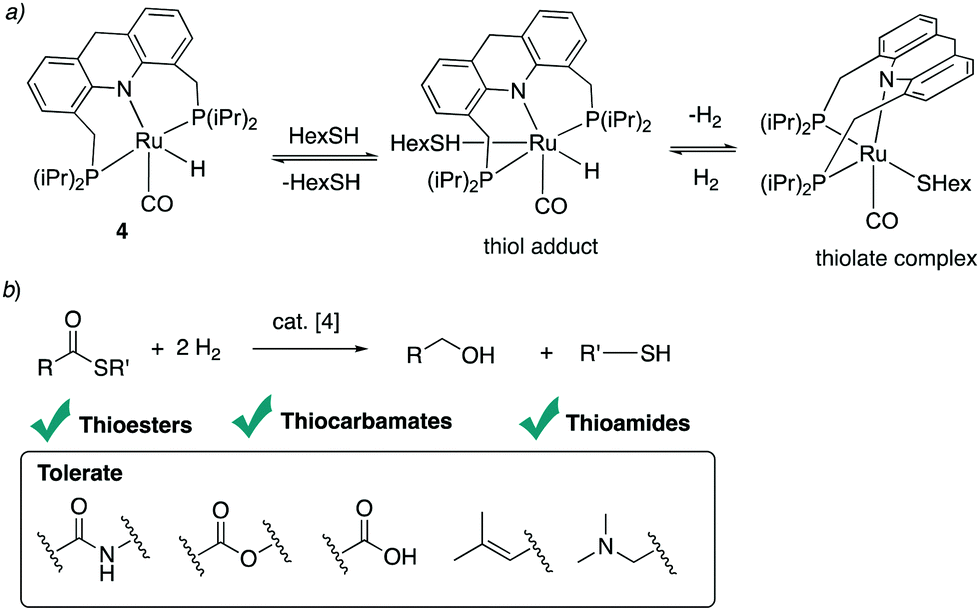
4.4.2. Thioester, thiocarbamate, thioamide hydrogenation.
In 2020 our group also reported the reverse reaction, namely thioester hydrogenation to alcohols and thiols under low H2 pressure, catalysed by the acridine-based dearomatized complexes.96 Under H2 pressure, the equilibrium between the thiol and thiolate complex becomes tilted toward the formation of thiol, which allows the reaction to proceed in the opposite direction (Fig. 32a). Thioester reduction, while readily achieved in biological systems by the thioester reductase enzyme under benign conditions, had been synthetically limited prior to this study to the use of stoichiometric reducing agents. Using the Ru–acridine complex 4 (1 mol%), hydrogenation of several thioesters to the corresponding alcohol and thiol under 20 bar H2 pressure at 135 °C was demonstrated (Fig. 32b). Interestingly, under the conditions, the thioester can be selectively hydrogenated even in the presence of other reducible functional groups such as esters or amide, which are more challenging to hydrogenate. Using the same complex 4 the hydrogenation of thiocarbamates and thioamides under 40 bars of H2 pressure was achieved with high to excellent yields. The mechanism of the hydrogenation was studied computationally by Poater et al., who also found that the hydrogenation is likely to proceed from the fac isomer, with the coordinating thiolate ligand playing an essential role in H2 activation during the reaction.97
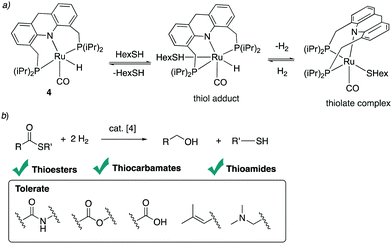 |
| | Fig. 32 (a) Interconversion between the thiol adduct and thiolate complex (b) hydrogenation of thioesters, thiocarbamates, and thioamides. | |
5. Summary and outlook
In this article we discussed how the acridine-based PNP pincer complexes differ from the traditional pincer complexes in structure and reactivity. Among some of the key points, the larger chelate size of the central acridine donor imparts flexibility on the whole system, resulting in possible hemilability with an elongated Ru–N bond distance (2.479 Å).53 Further, complexation with the N atom makes the 9CH position of the central acridine donor electrophilic and susceptible to hydride attack. Consequently, the aromatic acridine complexes can easily and irreversibly convert to the dearomatized (9CH2) PNP pincer complexes under various conditions. The dearomatized complexes are fluxional and can access the facial coordination mode during reaction. In case of Ru–dAcr(iPr) complex (4), the fac isomer is about 9 kcal mol−1 energetically uphill compared to the mer isomer,57 and the energy difference drastically decreases in the Ru–dAcr(Ph) system (∼5 kcal mol−1).59 The adoption of fac isomers enables a unique H2 liberation pathway from Ru–H and water, alcohols, thiols and acids, which is an initial elementary step in many reactions catalysed by these complexes (FA dehydrogenation, thioester formation and others). It also results in the rate enhancement that is often observed in the acridine-based PNP systems in presence of protic substrates such as water (fourfold for amine dehydrogenation56) or thiols (80 fold for methanol reforming91). It is surmised that after H2 liberation by reaction of water or thiol, the resulting hydroxy or thiolate complex can induce an outer-sphere hydride abstraction pathway which is more favourable than the inner sphere pathway. Another elemental step possible with the dearomatized acridine PNP system is beta hydride elimination due to the availability of two cis coordination sites for catalysis, which is crucial for many reactions, including amine dehydrogenation, formic acid dehydrogenation, enol ether to ester formation and others. Overall, the acridine based complexes catalyse several unique reactions, including conversion of alcohol to amine and vice versa, H2 generation from neat formic acid or water–methanol mixture, oxidation of furfural/5-hydroxymethyl furfural and cyclic amines with water, selective alcohol oxidation to acetals, which are not imitable by traditional pincer complexes involving pyridine based or aliphatic PNP type pincer complexes.
Among possible future developments with acridine-based pincer complexes, efforts can be directed toward developing first-row-transition metal complexes with acridine-based PNP pincer ligands. Many base metal pincer complexes with pyridine and aliphatic PNP ligands have been developed in recent years, which can show similar catalytic transformations of their noble metal counterparts. On the contrary, all reports involving acridine PNP pincer complexes have been limited to ruthenium. The difficulty is most likely due to the increased size of the acridine–PNP ligand, which requires a spacious second/third row transition metal for effective complexation. Notably, our group reported in 2013 an iron complex based on dearomatized–acriphos ligand and employed it for alkyne semi-hydrogenation to alkenes.98 Also in 2019, we reported a manganese dearomatized–acriphos complex for the ammonia-mediated conversion of alcohols to amides.99 However, synthesis of equivalent acridine–PNP complexes with these abundant first-row transition metals remains elusive. In another direction, the effect of electron density tuning on the ruthenium center of Ru–dAcr complexes has not been thoroughly investigated. We recently discovered that decreasing the electron density at the Ru centre, as with Ru–dAcr(Ph) complex 6, can significantly increase the selectivity of ester formation from enol ethers when compared to Ru–dAcr(iPr) complex 4.59 It will be interesting to see how further tuning of the phosphine substituents can affect the rate of elemental reactions in these systems via experimental investigation and DFT studies. Further, the presence of a spectator ligand, which has so far been a carbonyl group, provides another powerful opportunity to modify the electronic nature of these complexes to maximise catalytic activity and selectivity for a desired reaction. We hope that this report will encourage research into these fundamental directions in acridine-based PNP complex system, which, while superficially similar to its pyridine-based counterpart, appear to hide quite a few tricks up its sleeves that are yet to be explored.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This research is supported by the European Research Council (ERC AdG 692775). S. K. acknowledges the Sustainability and Energy Research Initiative (SAERI) of Weizmann Institute of Science for a research fellowship. D. M. holds the Israel Matz Professorial Chair of Organic Chemistry.
Notes and references
- M. E. van der Boom and D. Milstein, Chem. Rev., 2003, 103, 1759–1792 CrossRef CAS PubMed.
-
Organometallic Pincer Chemistry in Topics in Organometallic Chemistry, ed. G. van Koten, D. Milstein, Springer, Heidelberg, 2013, vol. 40 Search PubMed.
- E. Peris and R. H. Crabtree, Chem. Soc. Rev., 2018, 47, 1959–1968 RSC.
- M. A. W. Lawrence, K.-A. Green, P. N. Nelson and S. C. Lorraine, Polyhedron, 2018, 143, 11–27 CrossRef CAS.
- C. Gunanathan and D. Milstein, Chem. Rev., 2014, 114, 12024–12087 CrossRef CAS PubMed.
- S. Werkmeister, J. Neumann, K. Junge and M. Beller, Chem. – Eur. J., 2015, 21, 12226–12250 CrossRef CAS.
- G. van Koten, K. Timmer, J. G. Noltes and A. L. Spek, J. Chem. Soc., Chem. Commun., 1978, 250–252 RSC.
- C. J. Moulton and B. L. Shaw, J. Chem. Soc., Dalton Trans., 1976, 1020–1024 RSC.
- G. van Koten, Pure Appl. Chem., 1989, 61, 1681–1694 CAS.
- G. van Koten, T. K. Hollis and D. Morales-Morales, Eur. J. Inorg. Chem., 2020, 4416–4417 CrossRef CAS.
- A. Kumar, P. Daw and D. Milstein, Chem. Rev., 2022, 122, 385–441 CrossRef CAS PubMed.
- R. H. Crabtree, Chem. Rev., 2017, 117, 9228–9246 CrossRef CAS PubMed.
- K. Sordakis, C. Tang, L. K. Vogt, H. Junge, P. J. Dyson, M. Beller and G. Laurenczy, Chem. Rev., 2018, 118, 372–433 CrossRef CAS.
-
Green Catalysis, volume 1: Homogeneous Catalysis in Handbook of Green Chemistry, ed. P. T. Anastas and R. H. Crabtree, Wiley-VCH, 2014 Search PubMed.
- L. Piccirilli, D. Lobo Justo Pinheiro and M. Nielsen, Catalysts, 2020, 10, 773 CrossRef CAS.
-
C. Jensen, D. Brayton, S. W. Jorgensen and P. Hou, Development of a Practical Hydrogen Storage System Based on Liquid Organic Hydrogen Carriers and a Homogeneous Catalyst, Hawaii Hydrogen Carriers, LLC, Honolulu, HI, USA, 2017 Search PubMed.
- J.-Y. Cho, H. Kim, J.-E. Oh and B. Y. Park, Catalysts, 2021, 11, 1497 CrossRef CAS.
- J. Kothandaraman, S. Kar, R. Sen, A. Goeppert, G. A. Olah and G. K. S. Prakash, J. Am. Chem. Soc., 2017, 139, 2549–2552 CrossRef CAS.
- Y. Xie, P. Hu, Y. Ben-David and D. Milstein, Angew. Chem., Int. Ed., 2019, 58, 5105–5109 CrossRef CAS.
- S. Kar, J. Kothandaraman, A. Goeppert and G. K. S. Prakash, J. CO2 Util., 2018, 23, 212–218 CrossRef CAS.
- Y.-N. Li, R. Ma, L.-N. He and Z.-F. Diao, Catal. Sci. Technol., 2014, 4, 1498–1512 RSC.
- C. A. Huff and M. S. Sanford, J. Am. Chem. Soc., 2011, 133, 18122–18125 CrossRef CAS PubMed.
- S. Wesselbaum, T. vom Stein, J. Klankermayer and W. Leitner, Angew. Chem., 2012, 124, 7617–7620 CrossRef.
- S. Kar, A. Goeppert and G. K. S. Prakash, Acc. Chem. Res., 2019, 52, 2892–2903 CrossRef CAS PubMed.
- M. Nielsen, E. Alberico, W. Baumann, H.-J. Drexler, H. Junge, S. Gladiali and M. Beller, Nature, 2013, 495, 85–89 CrossRef CAS.
- R. E. Rodríguez-Lugo, M. Trincado, M. Vogt, F. Tewes, G. Santiso-Quinones and H. Grützmacher, Nat. Chem., 2013, 5, 342–347 CrossRef PubMed.
- J. Kothandaraman, S. Kar, A. Goeppert, R. Sen and G. K. S. Prakash, Top. Catal., 2018, 61, 542–559 CrossRef CAS.
- A. Kumar and C. Gao, ChemCatChem, 2021, 13, 1105–1134 CrossRef CAS.
- T. Ohkuma, H. Ooka, S. Hashiguchi, T. Ikariya and R. Noyori, J. Am. Chem. Soc., 1995, 117, 2675–2676 CrossRef CAS.
- T. Ohkuma, H. Ooka, T. Ikariya and R. Noyori, J. Am. Chem. Soc., 1995, 117, 10417–10418 CrossRef CAS.
- T. Ikariya, K. Murata and R. Noyori, Org. Biomol. Chem., 2006, 4, 393–406 RSC.
- M. R. Elsby and R. T. Baker, Chem. Soc. Rev., 2020, 49, 8933–8987 RSC.
- J. R. Khusnutdinova and D. Milstein, Angew. Chem., Int. Ed., 2015, 54, 12236–12273 CrossRef CAS PubMed.
- C. Gunanathan and D. Milstein, Acc. Chem. Res., 2011, 44, 588–602 CrossRef CAS.
- D. Milstein, Philos. Trans. R. Soc., A, 2015, 373, 20140189 CrossRef.
- T. P. Gonçalves, I. Dutta and K.-W. Huang, Chem. Commun., 2021, 57, 3070–3082 RSC.
- T. Shimbayashi and K. Fujita, Catalysts, 2020, 10, 635 CrossRef CAS.
- E. Fogler, J. A. Garg, P. Hu, G. Leitus, L. J. W. Shimon and D. Milstein, Chem. – Eur. J., 2014, 20, 15727–15731 CrossRef CAS.
- S. Kar, M. Rauch, A. Kumar, G. Leitus, Y. Ben-David and D. Milstein, ACS Catal., 2020, 10, 5511–5515 CrossRef CAS PubMed.
- S. Kar, Y. Xie, Q. Q. Zhou, Y. Diskin-Posner, Y. Ben-David and D. Milstein, ACS Catal., 2021, 11, 7383–7393 CrossRef CAS.
- A. Kumar, P. Daw, N. Angel Espinosa-Jalapa, G. Leitus, L. J. W. Shimon, Y. Ben-David and D. Milstein, Dalton Trans., 2019, 48, 14580–14584 RSC.
- K. Junge, V. Papa and M. Beller, Chem. – Eur. J., 2019, 25, 122–143 CrossRef CAS.
- A. Mukherjee, A. Nerush, G. Leitus, L. J. W. Shimon, Y. B. David, N. A. E. Jalapa and D. Milstein, J. Am. Chem. Soc., 2016, 138, 4298–4301 CrossRef CAS PubMed.
- T. Zell and D. Milstein, Acc. Chem. Res., 2015, 48, 1979–1994 CrossRef CAS PubMed.
- L. Alig, M. Fritz and S. Schneider, Chem. Rev., 2019, 119, 2681–2751 CrossRef CAS PubMed.
- S. Chakraborty, P. Bhattacharya, H. Dai and H. Guan, Acc. Chem. Res., 2015, 48, 1995–2003 CrossRef CAS PubMed.
- A. Awada, A. Moreno-Betancourt, C. Philouze, Y. Moreau, D. Jouvenot and F. Loiseau, Inorg. Chem., 2018, 57, 15430–15437 CrossRef CAS PubMed.
- S. J. Malthus, R. K. Wilson, D. S. Larsen and S. Brooker, Supramol. Chem., 2016, 28, 98–107 CrossRef CAS.
- O. Prakash, H. Joshi, U. Kumar, A. K. Sharma and A. K. Singh, Dalton Trans., 2014, 44, 1962–1968 RSC.
- S. Hillebrand, B. Bartkowska, J. Bruckmann, C. Krüger and M. W. Haenel, Tetrahedron Lett., 1998, 39, 813–816 CrossRef CAS.
- K. Rohmann, J. Kothe, M. W. Haenel, U. Englert, M. Hölscher and W. Leitner, Angew. Chem., Int. Ed., 2016, 55, 8966–8969 CrossRef CAS PubMed.
- C. Gunanathan and D. Milstein, Angew. Chem., Int. Ed., 2008, 47, 8661–8664 CrossRef CAS PubMed.
- C. Gunanathan, L. J. W. Shimon and D. Milstein, J. Am. Chem. Soc., 2009, 131, 3146–3147 CrossRef CAS PubMed.
- X. Ye, P. N. Plessow, M. K. Brinks, M. Schelwies, T. Schaub, F. Rominger, R. Paciello, M. Limbach and P. Hofmann, J. Am. Chem. Soc., 2014, 136, 5923–5929 CrossRef CAS.
- C. Gunanathan, B. Gnanaprakasam, M. A. Iron, L. J. W. Shimon and D. Milstein, J. Am. Chem. Soc., 2010, 132, 14763–14765 CrossRef CAS.
- S. Tang, M. Rauch, M. Montag, Y. Diskin-Posner, Y. Ben-David and D. Milstein, J. Am. Chem. Soc., 2020, 142, 20875–20882 CrossRef CAS PubMed.
- U. Gellrich, J. R. Khusnutdinova, G. M. Leitus and D. Milstein, J. Am. Chem. Soc., 2015, 137, 4851–4859 CrossRef CAS.
- J. Luo, M. Rauch, L. Avram, Y. Diskin-Posner, G. Shmul, Y. Ben-David and D. Milstein, Nat. Catal., 2020, 3, 887–892 CrossRef CAS.
- S. Kar, J. Luo, M. Rauch, Y. Diskin-Posner, Y. Ben-David and D. Milstein, Green Chem., 2022, 24, 1481–1487 RSC.
- E. Suárez, P. Plou, D. G. Gusev, M. Martín and E. Sola, Inorg. Chem., 2017, 56, 7190–7199 CrossRef.
- S. Imm, S. Bähn, L. Neubert, H. Neumann and M. Beller, Angew. Chem., Int. Ed., 2010, 49, 8126–8129 CrossRef CAS.
- D. Pingen, C. Müller and D. Vogt, Angew. Chem., Int. Ed., 2010, 49, 8130–8133 CrossRef CAS PubMed.
- J. R. Khusnutdinova, Y. Ben-David and D. Milstein, Angew. Chem., Int. Ed., 2013, 52, 6269–6272 CrossRef CAS PubMed.
- J. Zhang, G. Leitus, Y. Ben-David and D. Milstein, J. Am. Chem. Soc., 2005, 127, 10840–10841 CrossRef CAS PubMed.
- S. Chakraborty, P. O. Lagaditis, M. Förster, E. A. Bielinski, N. Hazari, M. C. Holthausen, W. D. Jones and S. Schneider, ACS Catal., 2014, 4, 3994–4003 CrossRef CAS.
- P. Daw, Y. Ben-David and D. Milstein, J. Am. Chem. Soc., 2018, 140, 11931–11934 CrossRef CAS.
- S. Michlik and R. Kempe, Nat. Chem., 2013, 5, 140–144 CrossRef CAS PubMed.
- M. Zhang, H. Neumann and M. Beller, Angew. Chem., Int. Ed., 2013, 52, 597–601 CrossRef CAS PubMed.
- M. Zhang, X. Fang, H. Neumann and M. Beller, J. Am. Chem. Soc., 2013, 135, 11384–11388 CrossRef CAS PubMed.
- S. Michlik and R. Kempe, Angew. Chem., Int. Ed., 2013, 52, 6326–6329 CrossRef CAS PubMed.
- Y. Xie, Y. Ben-David, L. J. W. Shimon and D. Milstein, J. Am. Chem. Soc., 2016, 138, 9077–9080 CrossRef CAS PubMed.
- K. Keitaro, M-u. Toyomi, O. Yasushi and I. Yasutaka, Chem. Lett., 2009, 38, 838–839 CrossRef.
- G. R. M. Dowson, M. F. Haddow, J. Lee, R. L. Wingad and D. F. Wass, Angew. Chem., Int. Ed., 2013, 52, 9005–9008 CrossRef CAS PubMed.
- S. Chakraborty, P. E. Piszel, C. E. Hayes, R. T. Baker and W. D. Jones, J. Am. Chem. Soc., 2015, 137, 14264–14267 CrossRef CAS PubMed.
- G. Xu, T. Lammens, Q. Liu, X. Wang, L. Dong, A. Caiazzo, N. Ashraf, J. Guan and X. Mu, Green Chem., 2014, 16, 3971–3977 RSC.
- N. V. Kulkarni, W. W. Brennessel and W. D. Jones, ACS Catal., 2018, 8, 997–1002 CrossRef CAS.
- S. Fu, Z. Shao, Y. Wang and Q. Liu, J. Am. Chem. Soc., 2017, 139, 11941–11948 CrossRef CAS PubMed.
- J. R. Khusnutdinova, Y. Ben-David and D. Milstein, J. Am. Chem. Soc., 2014, 136, 2998–3001 CrossRef CAS PubMed.
- S. Tang, Y. Ben-David and D. Milstein, J. Am. Chem. Soc., 2020, 142, 5980–5984 CrossRef CAS PubMed.
- S. Kar, Q.-Q. Zhou, Y. Ben-David and D. Milstein, J. Am. Chem. Soc., 2022, 144, 1288–1295 CrossRef CAS PubMed.
- R. Bielski and G. Grynkiewicz, Green Chem., 2021, 23, 7458–7487 RSC.
- R. Padilla, S. Koranchalil and M. Nielsen, Catalysts, 2021, 11, 1371 CrossRef CAS.
- Y.-Q. Zou, N. von Wolff, A. Anaby, Y. Xie and D. Milstein, Nat. Catal., 2019, 2, 415–422 CrossRef CAS PubMed.
- Q.-Q. Zhou, Y.-Q. Zou, Y. Ben-David and D. Milstein, Chem. – Eur. J., 2020, 26, 15487–15490 CrossRef CAS PubMed.
- S. Kar, M. Rauch, G. Leitus, Y. Ben-David and D. Milstein, Nat. Catal., 2021, 4, 193–201 CrossRef CAS.
- J. Eppinger and K.-W. Huang, ACS Energy Lett., 2017, 2, 188–195 CrossRef CAS.
- C. Guan, Y. Pan, T. Zhang, M. J. Ajitha and K.-W. Huang, Chem. – Asian J., 2020, 15, 937–946 CrossRef CAS PubMed.
- J. J. A. Celaje, Z. Lu, E. A. Kedzie, N. J. Terrile, J. N. Lo and T. J. Williams, Nat. Commun., 2016, 7, 11308 CrossRef CAS PubMed.
- S. Wang, H. Huang, T. Roisnel, C. Bruneau and C. Fischmeister, ChemSusChem, 2019, 12, 179–184 CrossRef CAS PubMed.
- S. Cohen, V. Borin, I. Schapiro, S. Musa, S. De-Botton, N. V. Belkova and D. Gelman, ACS Catal., 2017, 7, 8139–8146 CrossRef CAS.
- J. Luo, S. Kar, M. Rauch, M. Montag, Y. Ben-David and D. Milstein, J. Am. Chem. Soc., 2021, 143, 17284–17291 CrossRef CAS PubMed.
- A. Monney, E. Barsch, P. Sponholz, H. Junge, R. Ludwig and M. Beller, Chem.
Commun., 2014, 50, 707–709 RSC.
- E. A. Bielinski, M. Förster, Y. Zhang, W. H. Bernskoetter, N. Hazari and M. C. Holthausen, ACS Catal., 2015, 5, 2404–2415 CrossRef CAS.
- Y.-L. Zhan, Y.-B. Shen, S.-P. Li, B.-H. Yue and X.-C. Zhou, Chin. Chem. Lett., 2017, 28, 1353–1357 CrossRef CAS.
- M. Rauch, J. Luo, L. Avram, Y. Ben-David and D. Milstein, ACS Catal., 2021, 11, 2795–2807 CrossRef CAS.
- J. Luo, M. Rauch, L. Avram, Y. Ben-David and D. Milstein, J. Am. Chem. Soc., 2020, 142, 21628–21633 CrossRef CAS PubMed.
- M. Tomasini, J. Duran, S. Simon, L. M. Azofra and A. Poater, Mol. Catal., 2021, 510, 111692 CrossRef CAS.
- D. Srimani, Y. Diskin-Posner, Y. Ben-David and D. Milstein, Angew. Chem., Int. Ed., 2013, 52, 14131–14134 CrossRef CAS PubMed.
- P. Daw, A. Kumar, N. A. Espinosa-Jalapa, Y. Ben-David and D. Milstein, J. Am. Chem. Soc., 2019, 141, 12202–12206 CrossRef CAS PubMed.
|
| This journal is © The Royal Society of Chemistry 2022 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence and
David
Milstein
and
David
Milstein