
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis and redox activity of carbene-coordinated group 13 metal radicals†
Bin
Li
 a,
Blaise L.
Geoghegan
ab,
Hanns M.
Weinert
a,
Christoph
Wölper
a,
George E.
Cutsail
III
ab and
Stephan
Schulz
*ac
a,
Blaise L.
Geoghegan
ab,
Hanns M.
Weinert
a,
Christoph
Wölper
a,
George E.
Cutsail
III
ab and
Stephan
Schulz
*ac
aInstitute of Inorganic Chemistry, University of Duisburg-Essen, Essen 45117, Germany. E-mail: stephan.schulz@uni-due.de
bMax Planck Institute for Chemical Energy Conversion (CEC), Stiftstraße 34–36, Mülheim an der Ruhr 45470, Germany. E-mail: george.cutsail@cec.mpg.de
cCenter for Nanointegration Duisburg-Essen (Cenide), University of Duisburg-Essen, Duisburg 47057, Germany
First published on 10th March 2022
Abstract
Carbenes are known to stabilize main group element compounds with unusual electronic properties. Herein, we report the synthesis of carbene-stabilized group 13 metal radicals (cAAC)MX2(IPr) (M = Al, X = Br 3; M = Ga, X = Cl 4) and the corresponding cations [(cAAC)MX2(IPr)][B(C6F5)4] (M = Al, X = Br 5; M = Ga, X = Cl 6), which were characterized spectroscopically and by sc-XRD. Quantum chemical calculation gave insights into their electronic structures.
Radicals are important species in organic synthesis,1 polymer2 and plasma chemistry3 and in bioorganic chemistry.4 They typically tend to dimerize, hence kinetic stabilization by use of sterically demanding ligands or electronic stabilization by delocalization of the unpaired electron are major stabilizing strategies. In recent years, main group element radicals received increasing interest due to their unique physical and chemical properties,5 and several bulky and/or π-conjugated ligands were established which allowed for the isolation of electronically and sterically stabilized reactive radicals.
In contrast to widely known boron-based radicals,5f the number of heavier group 13 metal radicals is limited (Scheme 1) to dinuclear radical anions (I),6,7 in which the unpaired electron is localized in the πM–M orbital, mononuclear radical anions (II),8 cyclic Al biradical (III)9 and boryl-substituted neutral radicals (V, M = Ga–Tl).10 Recently, the N-heterocyclic carbene (NHC) coordinated radical cation IV was synthesized by one-electron oxidation reaction of the digallene.11 Cyclic (alkyl)(amino)carbenes (cAAC) are also known to stabilize main group metal radicals12 including group 13 metal radicals (VI,13a,bVII),13c but reactivity studies of such radicals are yet still missing.
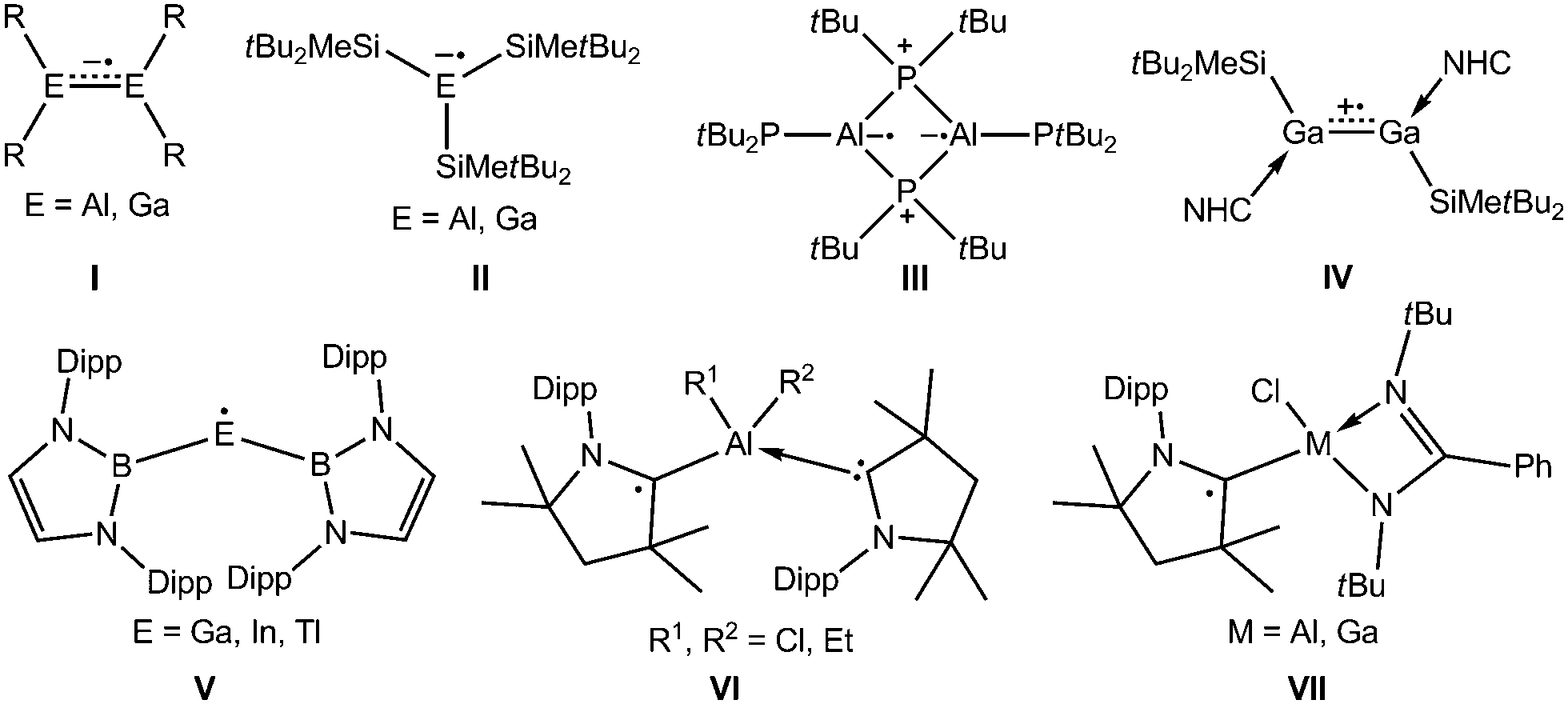 | ||
| Scheme 1 Examples of heavier group 13 elements containing radicals. R = CH(SiMe3)2, 2,4,6-iPr3C6H2; Dipp = 2,6-iPrC6H3. | ||
In view of the splendid performance of NHCs in stabilizing low-valent main group compounds14 and our general interest in main group metal radicals, we became interested in the synthesis and reactivity of group 13 metal radicals stabilized by one cAAC and one NHC, and we herein report on the syntheses of cAAC(IPr)MX2 radicals and their oxidation reactions.
Reduction of adducts (cAAC)MX3 (M = Al, X = Br, 1; M = Ga, X = Cl, 2; cAAC = C(Me)2CH2C(Me)2N(Dipp)C:; Dipp = 2,6-iPrC6H3) with two equivalents of KC8 in the presence of 1 equivalent of IPr (IPr = [C(Me)N(iPr)]2C:) gave red crystals in 26% (3) and 32% (4) yields after workup (Scheme 2), while only mixtures of the starting reagents and 3 and 4 were obtained from equimolar reactions. 3 and 4 are air and moisture sensitive solids, which are soluble in toluene and n-hexane. They are stable at ambient temperature for several months, whereas they decompose upon heating to 131 °C and 124 °C, respectively.
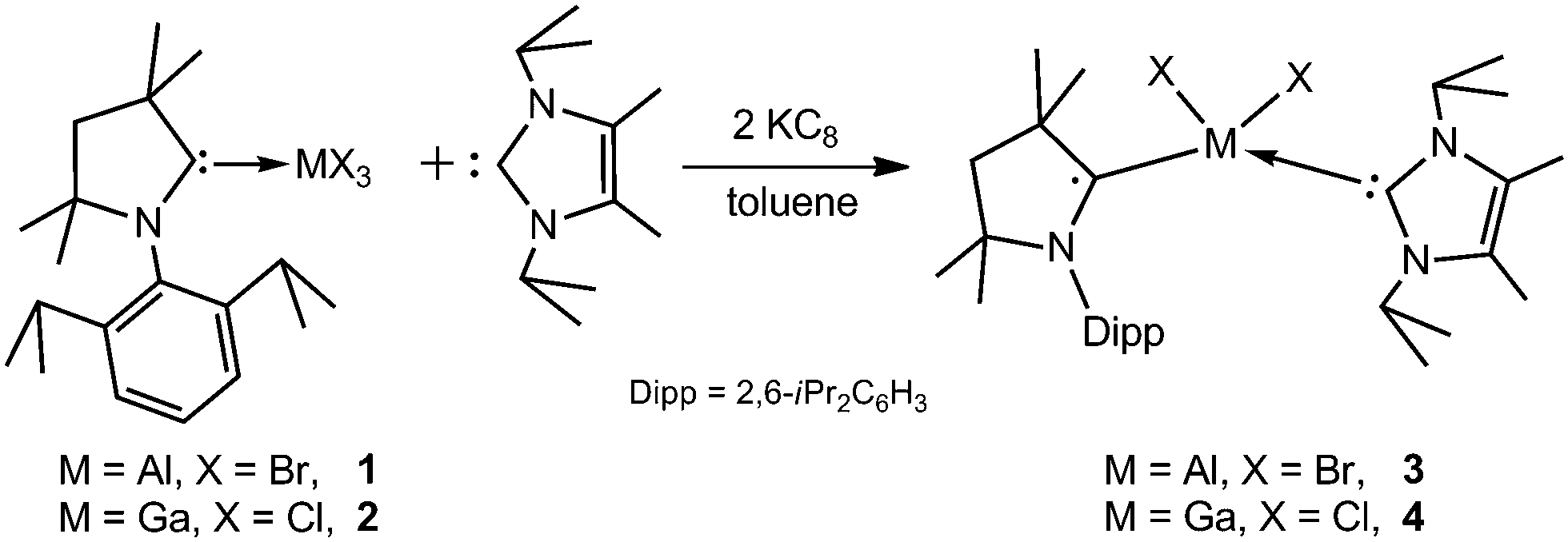 | ||
| Scheme 2 Synthesis of radical species 3 and 4. | ||
Single crystals of 3 (Fig. S16, ESI†) and 4 (Fig. 1) were obtained from concentrated toluene solutions upon storage at 4 °C. Both compounds crystallize in the monoclinic space group P21/c.
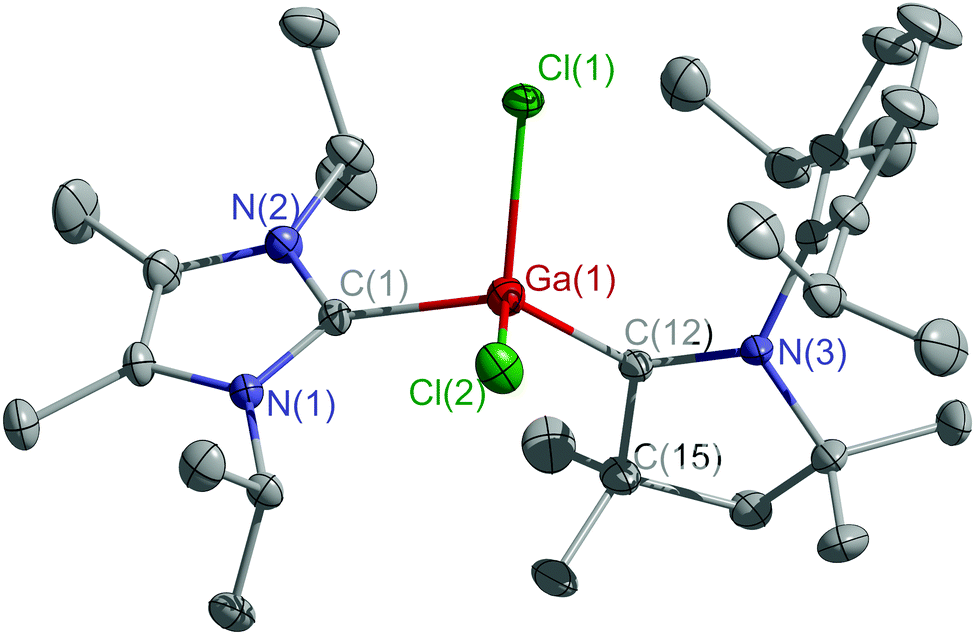 | ||
| Fig. 1 Molecular structure of 4. Thermal ellipsoids are drawn at 30% probability level. All hydrogen atoms are omitted for clarity. | ||
The C–M–C bond angles (3 113.20(5)°, 4 116.78(13)°) are wider than the Br–Al–Br (101.08(2)°) and Cl–Ga–Cl (100.68(5)°) bond angles (Table S2, ESI†). The Al–CcAAC bond length in 3 (1.941(1) Å) is typical for an Al–C σ-bond and comparable to other cAAC-coordinated aluminum radicals (VI, VII).13 The Al–CIPr bond length is far longer (2.078(1) Å) but similar to that reported for (IPr)Al(SitBu2Me)Br2 (2.062(3) Å),15 while the Ga–CcAAC bond length in 4 (1.932(3) Å) is shorter than the Ga–CIPr bond length (2.059(3) Å) and that in 2 (2.039(2) Å).16 Similar distances were found in (IPr)GaCl3 (2.011(4) Å),17 (IPr)Ga(Mes)Cl2 (1.978(2) Å)18 and (IPr)GaCl2(cAACH) (2.065(15) Å).19
Reactions of radicals 3 and 4 with [Ph3C][B(C6F5)4] yielded the cationic species 5 and 6 (Scheme 3). Although [R2ML2]+ cations (R = H, alkyl) are well known, knowledge of group 13 metal dihalide cations are still in their infancy. Compound 6 is soluble in polar solvents (THF, CH2Cl2), while 5 decomposes with formation of [cAACH][B(C6F5)4] (Fig. S19, ESI†). Cyclic voltammetry (CV) analyses demonstrate the redox reversibility between neutral radicals and cations (Fig. S23, ESI†). 1H NMR spectra of 5 and 6 show characteristic signals of cAAC and IPr ligands. The singlets at 2.36 (5) and 2.34 ppm (6) are assigned to the methyl groups on the IPr backbone, while the doublet at 1.54 ppm (5, 6) and the septet at 5.40 (5) and 5.27 ppm (6) are assigned to the isopropyl groups of IPr. In addition, two doublets (1.38, 1.39 ppm 5; 1.35, 1.38 ppm 6), one septet (2.74 (5), 2.70 (6) ppm) and three singlets (1.47, 1.56, 2.15 ppm 5; 1.48, 1.53, 2.20 ppm 6) are assigned to cAAC. The 13C NMR spectrum of 6 shows resonances of both carbene carbon atoms at 153.0 and 225.1 ppm.
 | ||
| Scheme 3 Synthesis of 5 and 6. | ||
Single crystals of 5 and 6 were obtained by layering n-hexane on the top of fluorobenzene solutions at ambient temperature. Compounds 5 (Fig. S18, ESI†) and 6 (Fig. 2) crystallize in the monoclinic space group P21/n and P21/c, respectively. The coordination geometry of cations 5 and 6 and radicals 3 and 4 is similar. The C–M–C bond angles (117.12(5)° 5, 117.59(5)° 6) are slightly wider than those of radicals 3 and 4, while the Al–CcAAC bond in cation 5 is elongated compared to that of 3, but close to those of adducts (cAAC)AlX3 (X = Cl, 2.037(1) Å; X = I, 2.049(2) Å).13b,20 Comparable findings were observed for cation 6, showing a longer Ga–CcAAC bond (2.037(1) Å) than radical 4 (1.932(3) Å), whereas comparable bond lengths were reported for (cAAC)GaCl3 (2.039(2) Å),16 (cAAC)GaHCl2 (2.053(2) Å),19 and (cAAC)2Ga2Cl4 (2.078(2) Å), respectively.21 The M–CIPr bonds (2.047(1) 5; 2.026(1) Å 6) are slightly shorter than those of neutral radicals 3 (2.078(1) Å) and 4 (2.059(3) Å), but still fall in the typical range of Al–CIPr and Ga–CIPr bond lengths.11,15,17–20
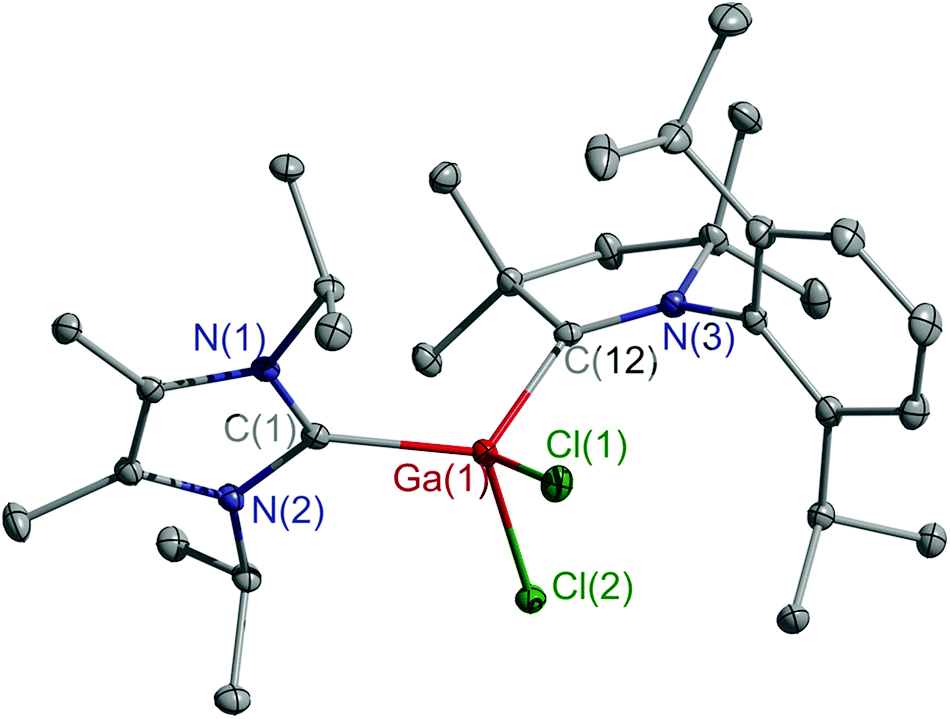 | ||
| Fig. 2 Molecular structure of 6. Thermal ellipsoids are drawn at 30% probability level. All hydrogen atoms and the anionic part are omitted for clarity. | ||
Electron paramagnetic resonance (EPR) spectroscopy was conducted on 3 and 4 to provide further insight into their electronic properties. The frozen solution (77 K) spectrum of 3 exhibits an isotropic S = 1/2 signal centred near ge = 2.0023 (Fig. 3). The spectrum of 3 is split by an isotropic 27Al (100% nat. abund.) hyperfine constant (HFC) of aiso(27Al) = 38 MHz, much smaller than the Al centred unpaired spin (A(27Al) = ∼1000 MHz),22 but more consistent with Al-cAAC radical species VI (aiso(27Al) = 14.6–23.4 MHz) where the unpaired electron is localized to the cAAC ligand.13a,b The room-temperature EPR of 3 (Fig. S22, ESI†) exhibits a similar broad isotropic spectrum to that of the 77 K spectrum, however, with some additional fine structure. Simulations of the room temperature spectrum estimate an Al aiso value of 35 MHz, in agreement with the low estimate from simulation of the 77 K spectrum (Fig. 3) and yielding a rough error estimate of ±5 MHz for the 77 K measurement. Additionally, a small 14N hyperfine coupling of 25 MHz is estimated, consistent with the nitrogen hyperfine couplings observed in other cAAC centered radicals.14a,23
 | ||
| Fig. 3 EPR spectra of 3 (left) and 4 (right) at 77 K in toluene. Simulation parameters for 3: g = 2.0023, aiso(Al) = 38 MHz, line-width (peak-to-peak) = 27 Gauss. Simulation parameters for 4: g = 2.0025, aiso(71Ga) = 196 MHz, aiso(69Ga) = 154 MHz, line-width (peak-to-peak) = 17 Gauss. | ||
The frozen solution EPR spectrum of 4 also exhibits an isotropic S = 1/2 signal centred near to ge, consistent with a light-atom centred radical and unlike the significant g-shifts observed in related two-coordinate Ga centred radicals.10 The EPR spectrum of 4 is split into four dominant lines, where the two outermost lines are further split, arising from the two I = 3/2 isotopes of gallium (69Ga (60.1%) and 71Ga (39.9%)) which exhibit HFCs that scale by the nuclear gyromagnetic ratios gn(71Ga)/gn(69Ga) = 1.271.24 The hyperfine pattern is predominantly isotropic, with aiso(69Ga) = 154 MHz and simulation of the EPR spectrum with a fairly large 17 Gauss linewidth (peak-to-peak) satisfactorily reproduces the experimental spectrum. For both 3 and 4, the relatively small Al/Ga HFCs and lack of resolvable/significant anisotropic hyperfine contributions in the frozen solution EPR spectra indicate that the unpaired electron is not Al/Ga p orbital centred but rather localized on the cAAC ligand. The small isotropic Al/Ga hyperfine coupling observed rather arises from spin polarization through the C–Al/Ga bonds, where the radical is C centred. Additionally, no significant 14N hyperfine coupling is resolved, and inclusion of up to 25 MHz 14N coupling by simulation remains unresolved in the broad EPR lines of the experimental spectrum (Fig. S21, ESI†). The lack of a large 14N HFC further supports a C centred radical species in both 3 and 4.
The bonding and electronic structure of radicals 3 and 4 and the cationic congeners 5 and 6 was evaluated by quantum chemical calculations.25 Calculated bond lengths within the C–MX2–C skeleton (Table S2, ESI†) are in good agreement with the experimental values. The SOMO of the parent radicals correlates well to the LUMO of cations (Fig. S24, ESI†), in line with reversible reduction presented in CV. The DFT calculations at the TPSSh/def2-TZVP level of theory show that most of the spin density resides on the C atom of the coordinating cAAC ligand in both 3 (0.73) and 4 (0.76), with significant spin density present on the adjacent N atoms (0.18) (Fig. 4a and b). Only a small amount of the total spin density is located at the Al/Ga centres (0.02), which is in line with the small isotropic HFCs used to simulate the experimental spectra but in contrast with the homoleptic carbene-coordinated Al radical VI,14a,14b in which the spin density is distributed unsymmetrically on the two carbenes, but still more than 17% spin density is located on the carbene with the minor contribution. It is noted that although the total spin density at the nitrogen is much larger than at the Al or Ga atoms, the Al and Ga hyperfine interaction dominate the EPR spectra due, in part, to their larger nuclear spins (more transitions) and their larger isotropic hyperfine coupling parameters (a0[14N] = 1811 MHz, a0[27Al] = 3911 MHz, a0[69Ga] = 12210 MHz).24 The DFT calculated spin densities and larger a0 values for Ga and Al in comparison to N supports the exclusion of a large N HFC (>25 MHz) in the simulations of the experimental spectra (Fig. S21, ESI†). The magnetic orbitals for 3 and 4 are displayed in Fig. 4c and d, respectively, and show that electron density is localized to the cAAC moiety and is well represented by the CcAAC–N π*-orbitals.
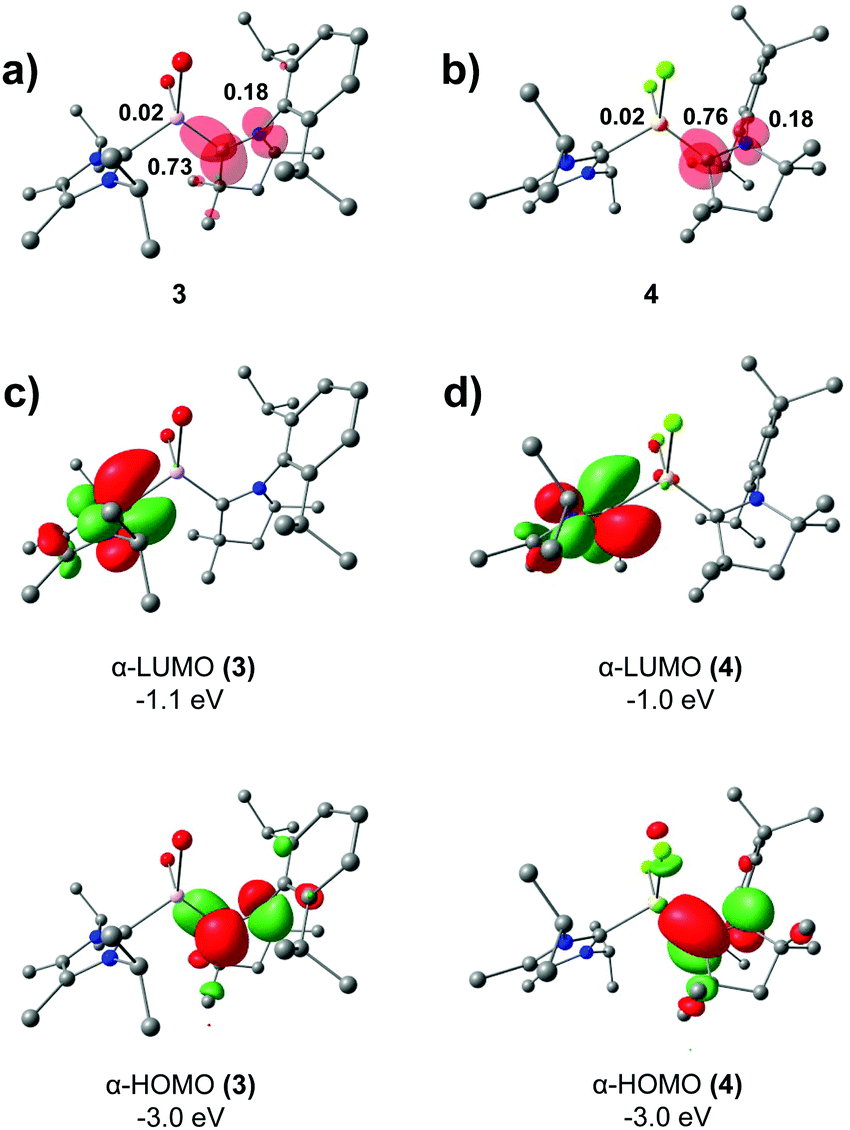 | ||
| Fig. 4 Spin density plots for (a) 3 and (b) 4 (isosurface value = 0.005 a.u.) with selected Mulliken spin population values. Selected Kohn–Sham MOs for (c) 3 and (d) 4 (isosurface value = 0.05 a.u.). | ||
Upon oxidation, one electron is removed from the CcAAC–N π*-orbital, resulting in increased WBIs (1.23 3, 1.23 4, 1.62 5, 1.63 6) and natural charges (–0.92 3, –0.87 4, –0.32 5, –0.28 6). In addition, the M–CcAAC bond is elongated in 5 and 6 despite the increasing charge, whereas the M–X and M–CIPr bonds become slightly shorter (WBI, Tables S3–S6, ESI†) as was observed in the solid state structures. Second order perturbation analysis of 3 and 4 showed a π-type interaction between a populated CcAAC-centred p-type orbital and the M–X σ* orbitals (3: 9.8 kcal mol−1; 4: 9.7 kcal mol−1) exclusively for the alpha spin orbitals, which is missing in the corresponding cations 5 and 6 and therefore explains the elongated CcAAC–M and shortened M–X bonds in 5 and 6. The change in charge upon oxidation also has only a negligible influence on the CcAAC–M bond length as the natural charge of the metal is almost unaffected (Tables S3–S6, ESI†) and the CcAAC–M bonds of 5 (2.0549(13) Å) and 6 (2.0367(12) Å) are almost identical with those of cAAC–AlBr3 (2.055(4) Å)26 and cAAC–GaCl3 (2.039(2) Å),16 respectively. The WBI of the CcAAC–Al bond is much lower compared to the CcAAC–Ga bond, which might explain the lower thermal stability of 5.
In conclusion, one electron oxidation of carbene-centred neutral radicals 3 and 4 gave cationic compounds 5 and 6, which are rare examples of carbene-coordinated group 13 cations.
Financial support by the University of Duisburg-Essen (S. S.) and the Max Planck Society (G. E. C.) is acknowledged. We thank Mrs J. Krüger for supporting quantum chemical calculations.
Conflicts of interest
There are no conflicts to declare.Notes and references
-
(a)
P. Renaud and M. P. Sibi, Radicals in Organic Synthesis, Wiley-VCH, 2001 CrossRef
; (b) N. Zhang, S. R. Samanta, B. M. Rosen and V. Percec, Chem. Rev., 2014, 114, 5848 CrossRef CAS PubMed
-
(a) K. Matyjaszewski and J. Xia, Chem. Rev., 2001, 101, 2921 CrossRef CAS PubMed
-
(a) J. S. Francisco, J. T. Muckerman and H. G. Yu, Acc. Chem. Res., 2010, 43, 1519 CrossRef CAS PubMed
-
(a)
C. Chatgilialoglu and A. Studer, Encyclopedia of Radicals in Chemistry, Biology and Materials, John Wiley & Sons Ltd, 2012, vol. 3 CrossRef
-
(a) I. Ratera and J. Veciana, Chem. Soc. Rev., 2012, 41, 303 RSC
-
(a) C. Pluta, K.-R. Pörschke, C. Krüger and K. Hildenbrand, Angew. Chem., Int. Ed. Engl., 1993, 32, 388 CrossRef
-
(a) X. He, R. A. Bartlett, M. M. Olmstead, K. Ruhlandt-Senge, B. E. Sturgeon and P. P. Power, Angew. Chem., Int. Ed. Engl., 1993, 32, 717 CrossRef
- M. Nakamoto, T. Yamasaki and A. Sekiguchi, J. Am. Chem. Soc., 2005, 127, 6954 CrossRef CAS PubMed
- P. Henke, T. Pankewitz, W. Klopper, F. Breher and H. Schnöckel, Angew. Chem., Int. Ed., 2009, 48, 8141 CrossRef CAS PubMed
- A. V. Protchenko, D. Dange, J. R. Harmer, C. Y. Tang, A. D. Schwarz, M.
J. Kelly, N. Phillips, R. Tirfoin, K. H. Birjkumar, C. Jones, N. Kaltsoyannis, P. Mountford and S. Aldridge, Nat. Chem., 2014, 6, 315 CrossRef CAS PubMed
- Z. Feng, Y. Fang, H. Ruan, Y. Zhao, G. Tan and X. Wang, Angew. Chem., Int. Ed., 2020, 59, 6769 CrossRef CAS PubMed
-
(a) M. Soleilhavoup and G. Bertrand, Acc. Chem. Res., 2015, 48, 256 CrossRef CAS PubMed
-
(a) S. Kundu, S. Sinhababu, S. Dutta, T. Mondal, D. Koley, B. Dittrich, B. Schwederski, W. Kaim, A. C. Stückl and H. W. Roesky, Chem. Commun., 2017, 53, 10516 RSC
-
(a) V. Nesterov, D. Reiter, P. Bag, P. Frisch, R. Holzner, A. Porzelt and S. Inoue, Chem. Rev., 2018, 118, 9678 CrossRef CAS PubMed
- P. Bag, A. Porzelt, P. J. Altmann and S. Inoue, J. Am. Chem. Soc., 2017, 139, 14384 CrossRef CAS PubMed
- A. El-Hellani, J. Monot, S. Tang, R. Guillot, C. Bour and V. Gandon, Inorg. Chem., 2013, 52, 11493 CrossRef CAS PubMed
- N. Marion, E. C. Escudero-Adán, J. Benet-Buchholz, E. D. Stevens, L. Fensterbank, M. Malacria and S. P. Nolan, Organometallics, 2007, 26, 3256 CrossRef CAS
- B. Quillian, P. Wei, C. S. Wannere, P. V. R. Schleyer and G. H. Robinson, J. Am. Chem. Soc., 2009, 131, 3169 CrossRef PubMed
- A. Hock, L. Werner, C. Luz and U. Radius, Dalton Trans., 2020, 49, 11108 RSC
- B. Li, S. Kundu, H. Zhu, H. Keil, R. Herbst-Irmer, D. Stalke, G. Frenking, D. M. Andrada and H. W. Roesky, Chem. Commun., 2017, 53, 2543 RSC
- J. K. Schuster, J. H. Muessig, R. D. Dewhurst and H. Braunschweig, Chem. – Eur. J., 2018, 24, 9692 CrossRef CAS PubMed
- B. Mile, J. A. Howard and J. S. Tee, Organometallics, 1988, 7, 1278 CrossRef CAS
-
(a) S. Roy, A. C. Stückl, S. Demeshko, B. Dittrich, J. Meyer, B. Maity, D. Koley, B. Schwederski, W. Kaim and H. W. Roesky, J. Am. Chem. Soc., 2015, 137, 4670 CrossRef CAS PubMed
-
J. A. Weil and J. R. Bolton, Electron paramagnetic resonance: elementary theory and practical applications, John Wiley & Sons, Hoboken, 2nd edn, 2007 Search PubMed
-
(a) F. Weigend, Phys. Chem. Chem. Phys., 2006, 8, 1057 RSC
- B. Li, CSD Commun., 2022 DOI:10.5517/ccdc.csd.cc2b7blj
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, spectroscopic characterization (NMR, EPR and IR spectra), cyclic voltammograms, elemental analysis and details of theoretical study of 3–6. For ESI and crystallographic data in CIF or other electronic format. CCDC 2129477 (3), 2129478 (4), 2129479 (5), 2129480 (5S) and 2129481 (6). For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d2cc00216g |
| This journal is © The Royal Society of Chemistry 2022 |