
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA mesoionic carbene complex of manganese in five oxidation states†
Benjamin
Wittwer‡
a,
Nicole
Dickmann‡
b,
Stephan
Berg‡
b,
Daniel
Leitner
a,
Lorenzo
Tesi
 c,
David
Hunger
c,
Raphael
Gratzl
a,
Joris
van Slageren
c,
Nicolas I.
Neuman
*de,
Dominik
Munz
*fg and
Stephan
Hohloch
*a
c,
David
Hunger
c,
Raphael
Gratzl
a,
Joris
van Slageren
c,
Nicolas I.
Neuman
*de,
Dominik
Munz
*fg and
Stephan
Hohloch
*a
aInstitute of General, Inorganic and Theoretical Chemistry, University of Innsbruck, Innrain 80-82, 6020 Innsbruck, Austria. E-mail: Stephan.Hohloch@uibk.ac.at
bUniversity of Paderborn, Faculty of Science, Department of Chemistry, Warburger Straße 100, 33098 Paderborn, Germany
cInstitute of Physical Chemistry, University of Stuttgart, Pfaffenwaldring 55, 70569 Stuttgart, Germany
dInstitute of Inorganic Chemistry, University of Stuttgart, Pfaffenwaldring 55, 70569 Stuttgart, Germany
eInstituto de Desarrollo Tecnológico para la Industria Química, INTEC, UNL-CONICET, Predio CONICET Santa Fe Dr Alberto Cassano, Ruta Nacional No 168, Km 0 Paraje El Pozo, (S3000ZAA) Santa Fe, Argentina. E-mail: nneuman@intec.unl.edu.ar
fInorganic Chemistry: Coordination Chemistry, Saarland University Campus C4 1, 66123 Saarbrücken, Germany. E-mail: Dominik.Munz@uni-saarland.de
gInorganic and General Chemistry, FAU Erlangen-Nürnberg, Egelandstr. 1, 91058 Erlangen, Germany
First published on 14th April 2022
Abstract
Reaction between a carbazole-based mesoionic carbene ligand and manganese(II) iodide results in the formation of a rare air-stable manganese(IV) complex after aerobic workup. Cyclic voltammetry reveals the complex to be stable in five oxidation states. The electronic structure of all five oxidation states is elucidated chemically, spectroscopically (NMR, high-frequency EPR, UV-Vis, MCD), magnetically, and computationally (DFT, CASSCF).
Pincer-ligands have not only enabled the development of various robust catalysts,1,2 and the stabilisation of highly reactive metal complexes,3 but they also opened the way to isolating metal centres in high oxidation states.4 However, pincers possessing an anionic5 CNC coordination motif with a donating amido moiety are still scarce and only a handful have been reported to date. Focusing on mesoionic carbenes6 (MICs), Bertrand and Bezuidenhout et al.7 recently reported a sterically encumbering CNC pincer with a carbazole backbone. The steric demand of this ligand allowed for the isolation of a series of highly reactive complexes such as a low-valent rhodium(I) oxygen adduct,8 or a T-shaped gold(I) complex,9 but it inhibited the formation of homoleptic metal complexes. However, given the propensity of NHC ligands to stabilize high-valent metal complexes,2,10 such as in a hexa-NHC-Mn(IV) complex by Smith and co-workers,11 the formation of homoleptic MIC complexes is a desirable goal. In our ongoing interest in mesoionic carbenes,12 we recently reported the synthesis and photophysical properties of a highly luminescent N-fused mesoionic carbene ligand and its s-block complexes13 as well as its catalytic potential in nickel(II) mediated CO2-epoxide coupling.14 Here we report its ability to stabilize high-valent manganese complexes.
The reaction between triazolium salt 1 and 0.5 equiv. of MnI2 with 3 equiv. of LiHMDS in THF afforded the deep green Mn(IV) complex 2(PF6)2 after aerobic workup (Fig. 1). First indication for the +IV oxidation state was given by the NMR Evans method, revealing an effective moment of 3.71 μB, suggesting the presence of three unpaired electrons, i.e. a d3 electron configuration, on the manganese ion.15 The assignment of the +IV oxidation state is further supported by the presence of two PF6 anions in its molecular X-ray structure (Fig. 1). The metal–carbene distances lie between 2.093(3) and 2.073(3) Å and are in the lower range of previously reported manganese NHC/MIC complexes.11,16 The nitrogen–manganese distances are 1.939(3) and 1.938(3) Å for Mn1–N10 and Mn1–N10A (Table S4, ESI†); comparable to previously reported Mn(IV) complexes.17,18 The UV-Vis spectrum of 2(PF6)2 shows a strong band at 750 nm in DCM, which shifts hypsochromically to 740 nm in acetone, supporting (in combination with calculations, vide infra) its assignment as an LMCT band (Fig. S10, ESI† further LMCTs are calculated to be in the range between 1000–1500 nm, cf. Fig. S27, ESI†). The EPR spectrum of solid 2(PF6)2 at 98 K (Fig. S11, ESI† left) shows a strongly anisotropic signal that could be simulated19 considering an S = 3/2 spin value and is consistent with previously reported Mn(IV) complexes.18 Simulation parameters are given in Table S2 (ESI†). In MeCN solution at 98 K the complex gives also a strongly anisotropic signal with g⊥ = 1.96 and g‖ = 2.08 (Fig. 2(b), see Table S2, ESI† for simulation parameters). The cyclic voltammogram of 2(PF6)2 in acetonitrile (Fig. 2(a)) revealed the presence of four reversible redox processes in solution. Upon comparison with the protonated ligand 1-I2 (Fig. S9, ESI†), the two reductions are most-likely metal-centred and correspond to the Mn(IV/III) (−0.55 V vs. Fc/[Fc]+) and Mn(III/II) (−2.02 V vs. Fc/[Fc]+) redox events. To set this into relation, Smith et al. reported reduction potentials of −0.71 V and −2.07 V vs. Fc/[Fc]+ for their Mn(IV/III) and Mn(III/II) hexa-NHC-manganese(IV) system.11 This indicates the carbazole ligand to be less donating. Further examples have been summarized in Table S1 (ESI†). Turning to the oxidative side, comparison of the redox potentials with those of related triazole complexes,20 as well as with the protonated ligand 1-I2 (Fig. S9, ESI†), indicates these processes to be ligand centred. Unfortunately in the present case, we failed to isolate any oxidized product from the reaction mixture using NOPF6 as oxidant due to fast re-reduction (less than 1 minute), which is a common observation with similar triazole complexes.20 Nevertheless, both oxidative states are accessible using SEC-EPR spectroscopy and were recorded in situ. Both oxidations give rise to paramagnetic species with well resolved EPR spectra. After the first oxidation, a sextet with g = 2.002 and mean aMn = 266 MHz is observed (Fig. S11, ESI† right). The second oxidation gives rise to a similar, but stronger signal, with a slightly larger hyperfine constant aMn = 290 MHz, and with each line experiencing a super-hyperfine interaction with two 14N nuclei, giving rise to a quintet with aN = 16 MHz (Fig. 2(c)). Both EPR spectra indicate ligand-centred oxidation processes in which the oxidized ligands couple antiferromagnetically to the d3 Mn(IV) centre resulting in an overall spin of S = 1 for [2]3+ and S = 1/2 for [2]4+ (vide infra). Moving to the reductive processes, chemical reduction of 2(PF6)2 in THF with 1 equiv. of KC8 resulted in the formation of the dark purple complex 2(PF6) (Fig. 1). X-ray structure analysis show the Mn–N distances to increase slightly, whereas the metal–carbene distances are only minorly affected by the redox processes. On the other hand, comparing intra-ligand distances in 2(PF6) with those in 1 and 2(PF6)2 reveals that these remain untouched by the reduction process. (Table S4, ESI†). The LMCT band at 750 nm in DCM decreases in intensity upon reduction of the metal centre, which supports a metal-based reduction process (Fig. S10, ESI†). Upon electrochemical reduction, the EPR signal of [2]2+ decreases, which is consistent with the formation of a Mn(III) species [2]+ with S = 1, which may be EPR silent at temperatures of 98 K and above. Unfortunately, all attempts to isolate the neutral complex [2] failed although numerous reducing agents, such as KC8, potassium metal, Na/Hg or Na/naphthalene were tried.
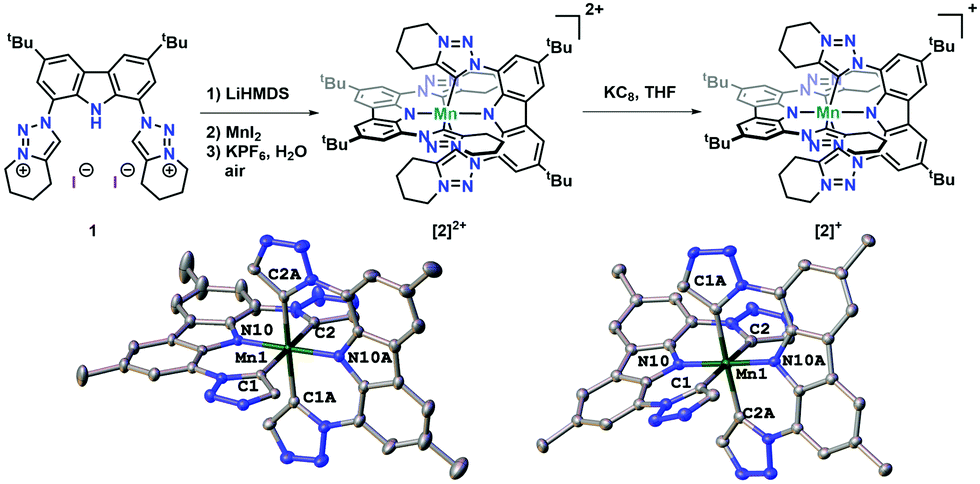 | ||
| Fig. 1 Synthesis of the Mn(IV) and Mn(III) complexes [2]2+ and [2]+ (top). Molecular structures of [2]2+ (bottom, left) and [2]+ (bottom, right). Counter ions, solvent molecules, tert-butyl and cyclohexyl residues have been omitted for clarity. | ||
 | ||
| Fig. 2 (a) CV of 2(PF6)2 in 0.1 M NBu4PF6 in MeCN at 298 K. Scan rate 100 mV s−1. The redox potentials vs. ferrocene from 2nd oxidation to 2nd reduction are as follows: 1.3 V, 0.78 V, −0.55 V and −2.02 V. (b) EPR spectra of [2]2+ in 0.1 M NBu4PF6 in MeCN at 98 K before electrolysis, and (c) after electrolysis at 1.93 V, corresponding to the second oxidized species [2]4+ at 283 K. Experimental EPR spectra are shown in black, whereas the simulations are shown in red. Please note that the small impurity (*) in the spectra of [2]4+ arises from non-electrolyzed [2]3+. (d) Curie plot of the experimental magnetic susceptibility of 2(PF6) and 2(PF6)2 powder samples measured in a static magnetic field of 10 kOe (1.8–300 K) for 2(PF6)2 and 1 kOe (1.8–50 K) and 10 kOe (40–300 K) for 2(PF6). Best fits are shown as solid lines. (e) High-frequency EPR spectra (solid black lines) and simulations (dashed red lines) of 2(PF6) at 10 K and various frequencies as indicated. The signal at g = 2 comes from an impurity in the sample holder. The grey solid lines are used to guide the eyes along the parallel and perpendicular directions, respectively. (f) MCD spectrum of 2(PF6) at 5 K and 5 T (black). The fit, based on a sum of Gaussian lines is shown as a red solid line, the single Gaussians as grey dashed-dotted lines. | ||
To further elucidate the electronic structure of the Mn(IV) and Mn(III) complexes, 2(PF6)2 and 2(PF6), respectively, direct current (dc) magnetometric measurements using SQUID magnetometry were performed (Fig. 2(d) and Fig. S12, ESI†). The measurements for 2(PF6)2 are in line with a putative Mn(IV) oxidation state (χMT = 1.88 cm3 K mol−1 at 300 K, ideally 1.875 cm3 K mol−1 for d3-configuration with S = 3/2 and g = 2). The simultaneous fit of χMT and magnetization curves at various temperatures (1.8, 5 and 10 K) using the same model applied to the X-Band EPR data and g-values (Table 1), provides zero-field splitting (ZFS) parameters (D = −0.42(1) cm−1 and E = −0.01(3) cm−1) in agreement with the EPR best-fit parameters. Magnetometry measurements of 2(PF6) result in a χMT value of 1.05 cm3 K mol−1 at 300 K, which points to a rare Mn(III) low-spin configuration (based on the Curie-Law, the ideal moment for d4 low-spin with g = 2 and S = 1 is χMT = 1 cm3 K mol−1). Furthermore, the simplest best-fit of χMT and magnetization curves (Fig. S12, ESI†) gives S = 1 with g = 2.0435(2) cm−1 and D = 12.75(1) cm−1. To confirm the presence of a Mn(III) low-spin complex, we recorded multi-frequency high-frequency EPR data of a powdered sample at 10 K, between 320 and 500 GHz (Fig. 2(e)). Simulations of the spectra assuming a S = 1 system provide an axial ZFS-term (D) value of 13.275(4) cm−1, with g⊥ = 2.00(1) and g‖ = 1.90(1), and a negligible transversal ZFS-term E, in agreement with the values obtained from magnetometry (vide supra). Magnetic circular dichroism (MCD) measurements were carried out on a frozen DCM/toluene solution sample of 2(PF6) in the visible region at 2 and 5 K in magnetic fields up to 5 T (Fig. 2(f)). The signal intensity of the MCD spectra increases at higher magnetic fields and lower temperatures (Fig. S15, ESI†), indicating C-term transitions. The MCD spectra show broad signals, that display a vibrational fine structure of 1100 cm−1 in the region of 14![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 500 to 18000 cm−1 (690 to 555 nm). According to TDDFT calculations, these signals are attributed to transitions with a mixed metal–ligand character (Table S11 and Fig. S35–S37, ESI†). Two broader features are observed at 19500 cm−1 and 23700 cm−1. The latter shows a negative MCD, which corresponds to a dominant absorption of right-hand circularly polarized light. Based on state-averaged CASSCF/NEVPT2 calculations (vide infra), we assign these three signals to mostly metal-based d–d transitions (Fig. S26, ESI†).
500 to 18000 cm−1 (690 to 555 nm). According to TDDFT calculations, these signals are attributed to transitions with a mixed metal–ligand character (Table S11 and Fig. S35–S37, ESI†). Two broader features are observed at 19500 cm−1 and 23700 cm−1. The latter shows a negative MCD, which corresponds to a dominant absorption of right-hand circularly polarized light. Based on state-averaged CASSCF/NEVPT2 calculations (vide infra), we assign these three signals to mostly metal-based d–d transitions (Fig. S26, ESI†).
| g | D | E | S | |
|---|---|---|---|---|
| *Frozen MeCN **solid sample ***see Fig. S11 (ESI) for further details. | ||||
| [2] | g ⊥ = n.a. (2.00) g‖ = n.a. (1.98) | n.a. | n.a. | 1/2 |
| [2]+ | SQUID: 2.0435(2) | SQUID: 12.75(1) cm−1 | 0 cm−1 (0.5 cm−1) | 1 |
| EPR: g⊥ = 2.00(1) (2.17); g‖ = 1.90(1) (1.97) | EPR: 13.275(4) cm−1 (22.0 cm−1) | |||
| [2]2+ | g ⊥ = 1.96* g‖ = 2.08* | −0.42(1) cm−1 (−3.0 cm−1) | −0.01(3) cm−1 (−0.4 cm−1) | |
| g ⊥ = 2.02** (2.00) g‖ = 1.93** (1.99) | 3/2 | |||
| [2]3+ | 2.002*** (2.002) | n.a. (4.06 cm−1) | n.a. (−0.27 cm−1) | 1 |
| [2]4+ | 2.00289 (2.00) | n.a. | n.a. | 1/2 |
To further shed light on the electronic structure of [2]+ and [2]2+ as well as metastable [2], [2]3+ and [2]4+, quantum chemical calculations were performed at the scalar relativistic DFT (ZORA-PBE; ZORA-PBE0; Fig. S20–S24, ESI†) and ZORA-CASSCF (Fig. S16–S19, ESI†) level of theory.21 The absorption spectra (Fig. S25–S34, ESI†) as well as the g-, D- and E-values calculated at the sa-CASSCF/NEVPT2 level of theory are in reasonable agreement with the experiment (Table 1). Cation [2]+ serves well to illustrate the electronic structures of the entire series. It features a low-spin, d4 configured (S = 1) metal ion, where the unpaired electrons occupy the degenerate d(xz) and d(yz) orbitals (Fig. 3(a)). Thus, and as expected for idealized D4h point group symmetry, a 1 + 2 + 1 + 1 ligand field splitting of the d-orbitals with an orbitally non-degenerate ground state is predicted. Reduction proceeds metal-based, affording [2] with an overall spin of S = 1/2 (Fig. S18, ESI†). Accordingly, metal-based oxidation removes one electron from the d(xy) orbital to give the high-spin (S = 3/2) manganese(IV) complex [2]2+ (Fig. S19, ESI† left). The oxidations to [2]3+ and [2]4+ (Fig. S19, ESI† right and Fig. 3(b)) proceed through stepwise removal of two electrons from the axial carbazolido π-donor ligands. Consequently, the metal remains in a formal oxidation state of +IV (vide supra). The weight of the lead configuration in [2]4+ amounts to c = 0.74. It relates to the electronic structure of a high-spin Mn(IV) ion antiferromagnetically coupled to the two unpaired electrons located at the two amido ligands.
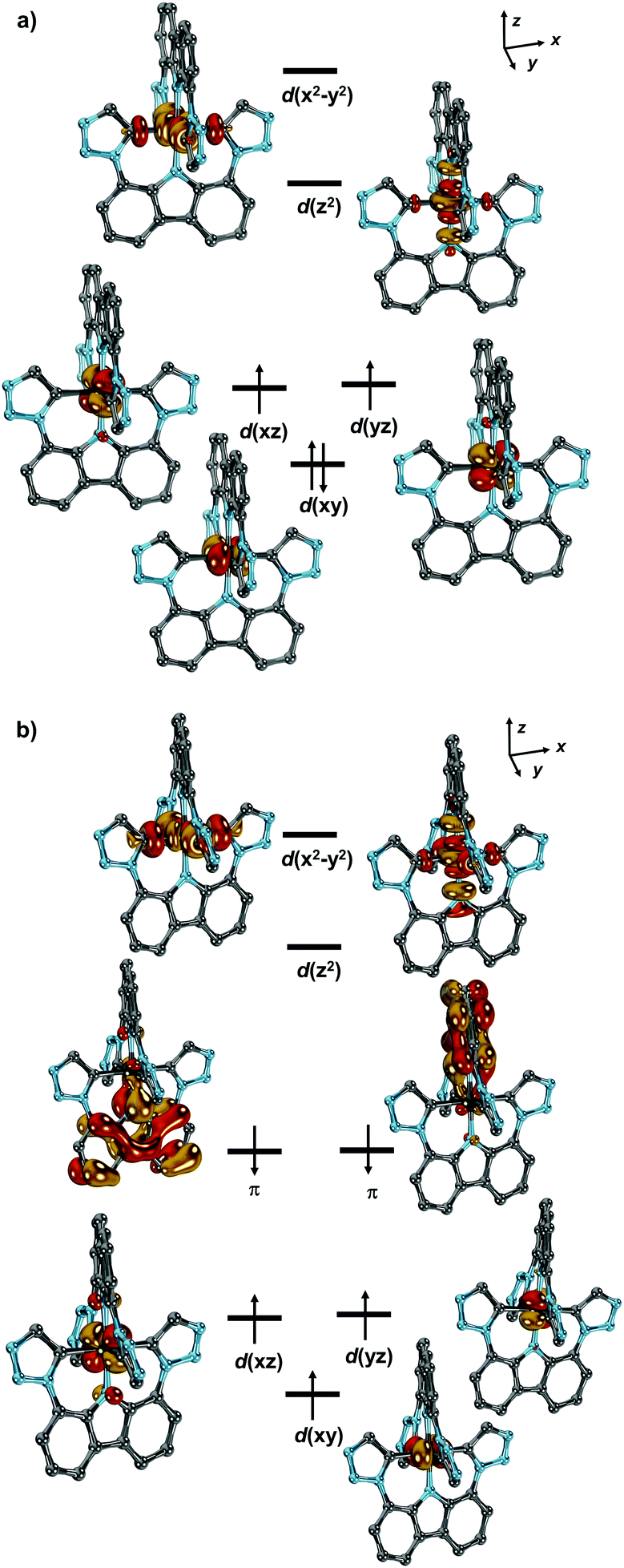 | ||
| Fig. 3 (a) Electronic structure of [2]+ according to sa-CASSCF(8,9) calculations (lead configuration c = 0.95); (b) electronic structure of [2]4+ according to sa-CASSCF(9,13) calculations (lead configuration: c = 0.74). Hydrogen atoms and N-fused MIC substituents have been omitted for clarity. | ||
In conclusion we have synthesized a rare example of an air-stable, high-valent Mn(IV) complex stabilized by 1,2,3-triazolylidene derived mesoionic carbene donors. The complex is electrochemically stable in five oxidation states, of which we were able to isolate and characterize the first reduced state, corresponding to a rare low-spin Mn(III) ion. The oxidized species are photosensitive and undergo quick reduction to the native state when exposed to light. CASSCF calculations revealed the oxidation to be ligand centred giving access to rare Mn(IV) organic radicals potentially interesting for photoredox catalysis. Further investigations in this direction are ongoing.
S. H. thanks the University of Innsbruck and Paderborn University for funding of this work. D. M. thanks the R. R. Z. Erlangen for computational resources. L. T., D. H. and J. v. S. thank the Baden-Württemberg Foundation (QTBW network, Moltriqusens project), the Landesgraduiertenförderung Baden-Württem-berg, and the DFG (SL104/10). N. I. N. thanks the University of Stuttgart for funding and is a member of CONICET.
Conflicts of interest
We declare no conflicts of interest.Notes and references
- (a) L. Alig, M. Fritz and S. Schneider, Chem. Rev., 2019, 119, 2681–2751 CrossRef CAS PubMed; (b) G. van Koten, J. Organomet. Chem., 2013, 730, 156–164 CrossRef CAS; (c) M. Baltrun, F. A. Watt, R. Schoch and S. Hohloch, Organometallics, 2019, 38, 3719–3729 CrossRef CAS.
- S. Liu, J. I. Amaro-Estrada, M. Baltrun, I. Douair, R. Schoch, L. Maron and S. Hohloch, Organometallics, 2021, 40, 107–118 CrossRef CAS.
- (a) J. Sun, J. Abbenseth, H. Verplancke, M. Diefenbach, B. de Bruin, D. Hunger, C. Würtele, J. van Slageren, M. C. Holthausen and S. Schneider, Nat. Chem., 2020, 12, 1054–1059 CrossRef CAS PubMed; (b) T. Kurogi, P. J. Carroll and D. J. Mindiola, J. Am. Chem. Soc., 2016, 138, 4306–4309 CrossRef CAS PubMed.
- (a) J. Abbenseth, M. Finger, C. Würtele, M. Kasanmascheff and S. Schneider, Inorg. Chem. Front., 2016, 3, 469–477 RSC; (b) A. J. Kosanovich, J. H. Reibenspies and O. V. Ozerov, Organometallics, 2016, 35, 513–519 CrossRef CAS.
- (a) S. Hameury, P. de Frémont and P. Braunstein, Chem. Soc. Rev., 2017, 46, 632–733 RSC; (b) S. T. Liddle, I. S. Edworthy and P. L. Arnold, Chem. Soc. Rev., 2007, 36, 1732–1744 RSC.
- (a) D. Schweinfurth, L. Hettmanczyk, L. Suntrup and B. Sarkar, Z. Anorg. Allg. Chem., 2017, 643, 554–584 CrossRef CAS; (b) G. Guisado-Barrios, M. Soleilhavoup and G. Bertrand, Acc. Chem. Res., 2018, 51, 3236–3244 CrossRef CAS PubMed; (c) G. Guisado-Barrios, J. Bouffard, B. Donnadieu and G. Bertrand, Angew. Chem., Int. Ed., 2010, 49, 4759–4762 CrossRef CAS PubMed; (d) P. Mathew, A. Neels and M. Albrecht, J. Am. Chem. Soc., 2008, 130, 13534–13535 CrossRef CAS PubMed.
- D. I. Bezuidenhout, G. Kleinhans, G. Guisado-Barrios, D. C. Liles, G. Ung and G. Bertrand, Chem. Commun., 2014, 50, 2431–2433 RSC.
- G. Kleinhans, G. Guisado-Barrios, D. C. Liles, G. Bertrand and D. I. Bezuidenhout, Chem. Commun., 2016, 52, 3504–3507 RSC.
- G. Kleinhans, M. M. Hansmann, G. Guisado-Barrios, D. C. Liles, G. Bertrand and D. I. Bezuidenhout, J. Am. Chem. Soc., 2016, 138, 15873–15876 CrossRef CAS PubMed.
- (a) M. Keilwerth, L. Grunwald, W. Mao, F. W. Heinemann, J. Sutter, E. Bill and K. Meyer, J. Am. Chem. Soc., 2021, 143, 1458–1465 CrossRef CAS PubMed; (b) J. L. Martinez, S. A. Lutz, H. Yang, J. Xie, J. Telser, B. M. Hoffman, V. Carta, M. Pink, Y. Losovyj and J. M. Smith, Science, 2020, 370, 356–359 CrossRef CAS PubMed; (c) D. Munz, Organometallics, 2018, 37, 275–289 CrossRef CAS; (d) C. F. Harris, M. B. Bayless, N. P. van Leest, Q. J. Bruch, B. N. Livesay, J. Bacsa, K. I. Hardcastle, M. P. Shores, B. de Bruin and J. D. Soper, Inorg. Chem., 2017, 56, 12421–12435 CrossRef CAS PubMed; (e) L. Gravogl, F. W. Heinemann, D. Munz and K. Meyer, Inorg. Chem., 2020, 59, 5632–5645 CrossRef CAS PubMed.
- A. P. Forshaw, R. P. Bontchev and J. M. Smith, Inorg. Chem., 2007, 46, 3792–3794 CrossRef CAS PubMed.
- (a) P. Dierks, A. Kruse, O. S. Bokareva, M. J. Al-Marri, J. Kalmbach, M. Baltrun, A. Neuba, R. Schoch, S. Hohloch, K. Heinze, M. Seitz, O. Kühn, S. Lochbrunner and M. Bauer, Chem. Commun., 2021, 57, 6640–6643 RSC; (b) M. Baltrun, F. A. Watt, R. Schoch, C. Wölper, A. G. Neuba and S. Hohloch, Dalton Trans., 2019, 48, 14611–14625 RSC; (c) S. Hohloch, L. Suntrup and B. Sarkar, Inorg. Chem. Front., 2016, 3, 67–77 RSC; (d) F. R. Neururer, S. Liu, D. Leitner, M. Baltrun, K. R. Fisher, H. Kopacka, K. Wurst, L. J. Daumann, D. Munz and S. Hohloch, Inorg. Chem., 2021, 60, 15421–15434 CrossRef CAS PubMed; (e) M. Rigo, L. Hettmanczyk, F. J. L. Heutz, S. Hohloch, M. Lutz, B. Sarkar and C. Müller, Dalton Trans., 2016, 46, 86–95 RSC.
- P. Pinter, C. M. Schüßlbauer, F. A. Watt, N. Dickmann, R. Herbst-Irmer, B. Morgenstern, A. Grünwald, T. Ullrich, M. Zimmer, S. Hohloch, D. M. Guldi and D. Munz, Chem. Sci., 2021, 12, 7401–7410 RSC.
- F. A. Watt, B. Sieland, N. Dickmann, R. Schoch, R. Herbst-Irmer, H. Ott, J. Paradies, D. Kuckling and S. Hohloch, Dalton Trans., 2021, 50, 17361–17371 RSC.
- (a) Z. S. Teweldemedhin, R. L. Fuller and M. Greenblatt, J. Chem. Educ., 1996, 73, 906 CrossRef CAS; (b) C. J. O’Connor, in Progress in inorganic chemistry. Volume 29, ed. S. J. Lippard, Wiley, New York, N.Y., 1982, vol. 453, pp. 203–283 Search PubMed; (c) R. L. Carlin, Magnetochemistry, Springer Berlin Heidelberg, Berlin, Heidelberg, 1986 CrossRef.
- (a) S. Friães, S. Realista, C. S. B. Gomes, P. N. Martinho, L. F. Veiros, M. Albrecht and B. Royo, Dalton Trans., 2021, 50, 5911–5920 RSC; (b) M. F. Pinto, M. Olivares, Á. Vivancos, G. Guisado-Barrios, M. Albrecht and B. Royo, Catal. Sci. Technol., 2019, 9, 2421–2425 RSC.
- (a) A. S. Attia and C. G. Pierpont, Inorg. Chem., 1998, 37, 3051–3056 CrossRef CAS; (b) H. Chun, P. Chaudhuri, T. Weyhermüller and K. Wieghardt, Inorg. Chem., 2002, 41, 790–795 CrossRef CAS PubMed; (c) S. K. Larsen and C. G. Pierpont, J. Am. Chem. Soc., 1988, 110, 1827–1832 CrossRef CAS.
- N. Leconte, J. Moutet, K. Herasymchuk, R. M. Clarke, C. Philouze, D. Luneau, T. Storr and F. Thomas, Chem. Commun., 2017, 53, 2764–2767 RSC.
- S. Stoll and A. Schweiger, J. Magn. Reson., 2006, 178, 42–55 CrossRef CAS PubMed.
- I. Pryjomska-Ray, D. Zornik, M. Pätzel, K. B. Krause, L. Grubert, B. Braun-Cula, S. Hecht and C. Limberg, Chem. – Eur. J., 2018, 24, 5341–5349 CrossRef CAS PubMed.
- (a) F. Neese, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2012, 2, 73–78 CAS; (b) F. Neese, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2018, 8, e1327 Search PubMed; (c) S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed; (d) S. Grimme, S. Ehrlich and L. Goerigk, J. Comput. Chem., 2011, 32, 1456–1465 CrossRef CAS PubMed; (e) C. Angeli, R. Cimiraglia, S. Evangelisti, T. Leininger and J.-P. Malrieu, J. Chem. Phys., 2001, 114, 10252–10264 CrossRef CAS; (f) C. Adamo and V. Barone, J. Chem. Phys., 1999, 110, 6158–6170 CrossRef CAS; (g) B. A. Heß, C. M. Marian, U. Wahlgren and O. Gropen, Chem. Phys. Lett., 1996, 251, 365–371 CrossRef; (h) R. Izsák and F. Neese, J. Chem. Phys., 2011, 135, 144105 CrossRef PubMed; (i) G. Knizia and J. E. M. N. Klein, Angew. Chem., Int. Ed., 2015, 54, 5518–5522 CrossRef CAS PubMed; (j) F. Neese, F. Wennmohs, A. Hansen and U. Becker, Chem. Phys., 2009, 356, 98–109 CrossRef CAS; (k) D. A. Pantazis, X.-Y. Chen, C. R. Landis and F. Neese, J. Chem. Theory Comput., 2008, 4, 908–919 CrossRef CAS PubMed; (l) J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed; (m) B. O. Roos, P. R. Taylor and P. E. Sigbahn, Chem. Phys., 1980, 48, 157–173 CrossRef CAS; (n) M.-C. Tseng, H.-T. Cheng, M.-J. Shen and Y.-H. Chu, Org. Lett., 2011, 13, 4434–4437 CrossRef CAS PubMed; (o) E. van Lenthe, E. J. Baerends and J. G. Snijders, J. Chem. Phys., 1993, 99, 4597–4610 CrossRef CAS; (p) C. van Wüllen, J. Chem. Phys., 1998, 109, 392–399 CrossRef; (q) F. Weigend, Phys. Chem. Chem. Phys., 2006, 8, 1057–1065 RSC; (r) F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 1953710 and 1993282. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2cc00097k |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2022 |