
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Expedient metal-free preparation of aryl aziridines via thermal cycloaddition reactions†
Filip
Sebest‡
 ,
Lalita
Radtanajiravong‡
,
Siim
Kaukver
,
Andrew J. P.
White
and
Silvia
Díez-González
*
,
Lalita
Radtanajiravong‡
,
Siim
Kaukver
,
Andrew J. P.
White
and
Silvia
Díez-González
*
Imperial College London, Department of Chemistry, MSRH, 82 Wood Lane, London W12 0BZ, UK. E-mail: s.diez-gonzalez@imperial.ac.uk
First published on 15th February 2022
Abstract
A straightforward synthesis of aryl aziridines is reported from readily available azides and alkenes and using technical solvents in the presence of air. This methodology does not require any additives and the obtained compounds can be employed in ring-opening and ring-expansion reactions.
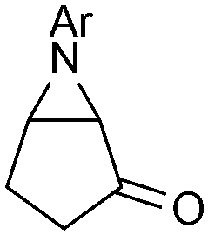
Aziridines, the smallest nitrogen-containing heterocycle, are found in biologically active natural products and are key intermediates in the synthesis of a wide array of nitrogenated derivatives.1 A range of methodologies have been developed for the stereoselective synthesis of these valuable heterocycles,2 but arguably metal-mediated transfer reactions (either of a nitrogen source to olefins or a carbon source to imines) remain the most prevalent options. Chemists’ fascination for this motif is far from waning and new, elegant alternative metal-free methodologies continue to appear in the literature,3 such as the hypoiodite mediated aziridination of alkenes with N-aminophthalimide,4 an aza-Prilezhaev reaction,5 or the electrochemical coupling of amines and alkenes,6 just to name a few (Scheme 1). While most of these reactions are limited to electron-rich alkenes, amine organocatalysis is a good alternative for α,β-unsaturated aldehydes.7 In this context, decades before azides gained their current popularity as nitrogen sources for the addition of the corresponding nitrenes to alkenes,8 pioneer work by Huisgen showed that electron-deficient alkenes deliver 1,4-disubstituted triazolines regioselectively when reacted with azides,9 which might deliver the corresponding aziridines upon nitrogen extrusion (Scheme 2).10 Subsequent reports of thermolysis11 or photolysis12 of triazolines to access aziridines have since emerged in the literature, however, these remain anecdotical in comparison to other methodologies due to selectivity issues. While the presence of electron-withdrawing substituents in triazoline 3 facilitates the cycloaddition step, it also increases the stability of its open-ring isomer that might evolve into aziridine 4, or a diazo compound 5. The latter can further react with an alkene to form after tautomerisation 2-pyrazoline 6,10,11 or undergo elimination to form an aminoalkane 7.13 The fate of these transient 1,4-disubstituted triazolines is strongly dependent on the substituents on either starting material and substantial decomposition is also often observed. More recently, the reaction of alkyl azides and alkenes with an electron-withdrawing group in the presence of an excess of triflic acid at low temperatures has been reported to produce aziridines in good to excellent yields.14 However, aryl azides only led to moderate yields due to azide decomposition under the optimal conditions.
 | ||
| Scheme 1 Selected metal-free syntheses of aziridines. PhthNH2 = N-aminophthalimide; TFE = tetrafluoroethylene; HfsNH2 = 1,1,1,3,3,3-hexafluoropropan-2-yl sulfamate. | ||
 | ||
| Scheme 2 Evolution of 1,4-disubstituted 1,2,3-triazolines. | ||
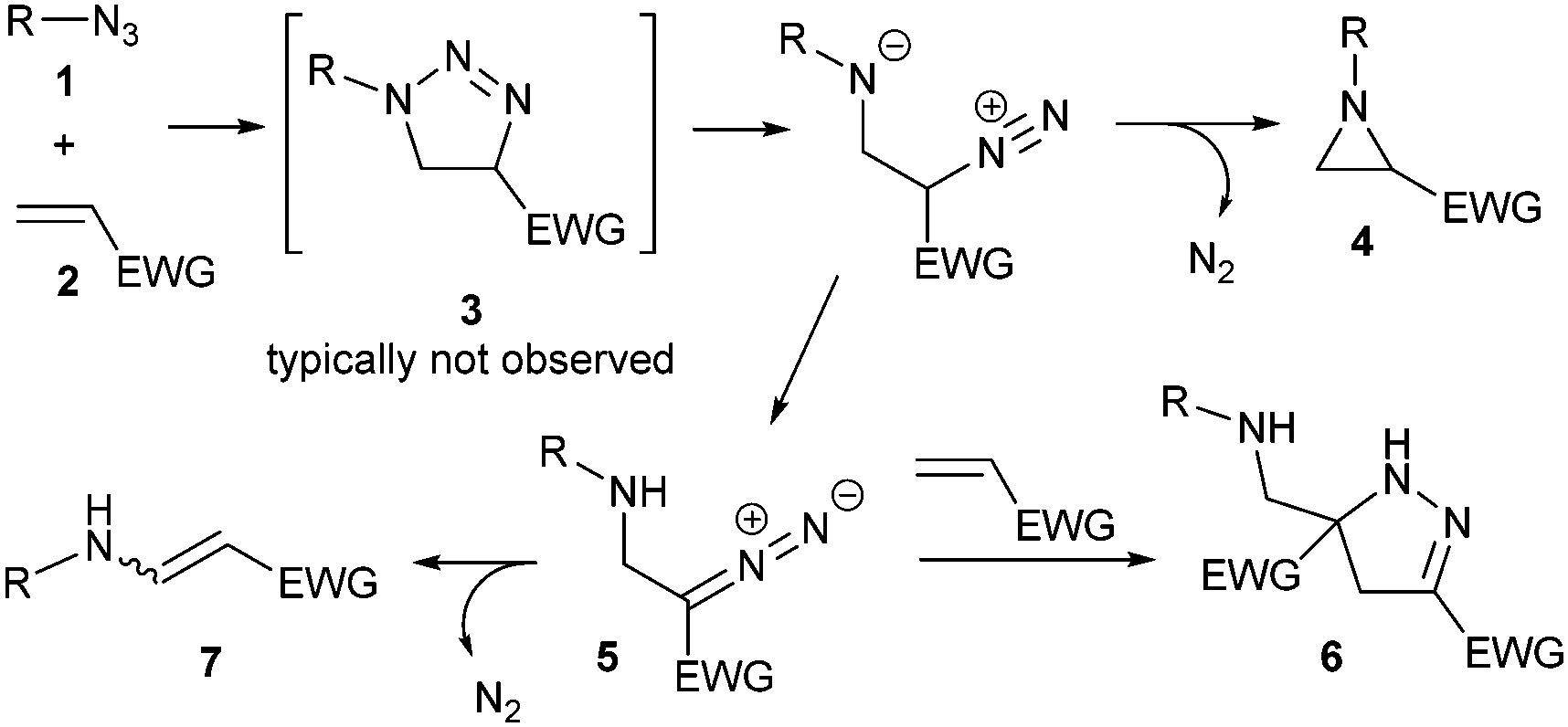
During our previous work on the preparation of pyrazolines from aryl azides and alkenes with an electron-withdrawing group,15 we noticed the significant formation of aziridines in some reactions involving ortho-substituted aryl azides. Since the formation of pyrazolines was facilitated by using DES (Deep Eutectic Solvent, choline chloride/urea 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2) as solvent, we tested alternative technical grade solvents with mesityl azide 1a and methyl acrylate 2a as model substrates (Table 1). The desired aziridine 4aa was the major product in all cases, and 1,5-disubstituted triazoline 3aa was the only other product identified in these reactions. Such product distribution indicates that all the originally formed 1,4-disubstituted triazoline evolved selectively into the corresponding aziridine. Remarkably, the reaction proceeded well even on water or in the absence of any solvent (Table 1, entries 7 and 8). Overall, either toluene or 1,4-dioxane provided the best results (Table 1, entries 1 and 2), and toluene was selected for further studies due to its relatively lower environmental impact.16
:2) as solvent, we tested alternative technical grade solvents with mesityl azide 1a and methyl acrylate 2a as model substrates (Table 1). The desired aziridine 4aa was the major product in all cases, and 1,5-disubstituted triazoline 3aa was the only other product identified in these reactions. Such product distribution indicates that all the originally formed 1,4-disubstituted triazoline evolved selectively into the corresponding aziridine. Remarkably, the reaction proceeded well even on water or in the absence of any solvent (Table 1, entries 7 and 8). Overall, either toluene or 1,4-dioxane provided the best results (Table 1, entries 1 and 2), and toluene was selected for further studies due to its relatively lower environmental impact.16
|
|
|||
|---|---|---|---|
| Entry | Solvent | 4aa (%)b | 3aa (%)b |
| a Reaction conditions: Azide 1a (1 mmol), alkene 2a (2.2 mmol) in 2 mL of solvent. b 1H NMR yields are the average of two independent experiments and were determined with respect to 1,3,5-trimethoxybenzene as internal standard. | |||
| 1 | Toluene | 90 | 10 |
| 2 | 1,4-Dioxane | 92 | 8 |
| 3 | THF | 75 | 11 |
| 4 | EtOAc | 84 | 12 |
| 5 | EtOH | 71 | 12 |
| 6 | Brine | 65 | 12 |
| 7 | Water | 73 | — |
| 8 | Neat | 77 | 12 |

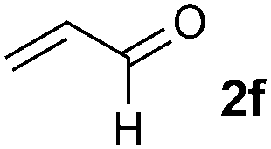
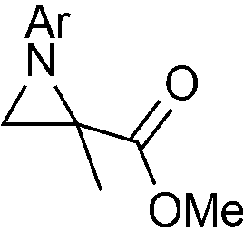
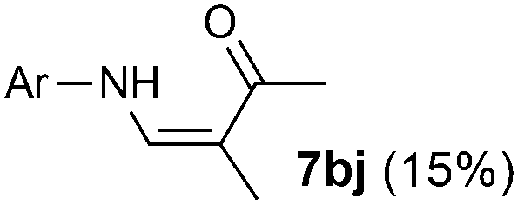
We then screened a range of mono- and di-substituted alkenes with 2-chlorophenyl azide 1b as model substrate (Table 2). The reaction tolerated a range of functional groups such as esters, amides, aldehydes and nitriles. Only in the case of diethyl vinyl phosphonate 2h a low yield was obtained and it could not be significantly improved by changing the reaction time or temperature (Table 2, entry 8). Pyrazoline formation was only detected with dimethyl fumarate 2o as starting alkene (Table 2, entry 15), but small quantities of aminoalkenes 7 were often observed. By-products 7 were obtained as Z-isomers only, as favoured by the formation of a hydrogen bond between the amine and the different carbonyl groups.
|
|
||||
|---|---|---|---|---|
| Entry | Alkene | 4bX (%) | Additional products | |
| a Reaction conditions: Azide 1b (2 mmol), alkene 2X (4 mmol) in 4 mL of technical toluene. b Isolated yields. 1H NMR yields are given in brackets and were calculated using 1,3,5-trimethoxybenzene as internal standard and are the average of at least two independent experiments. c 6.5 mmol of azide 1b. d 9.8 mmol of azide 1b. e 90 °C. f 110 °C. g 100 °C. | ||||
| 1c |
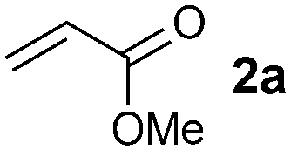
|

|
83 (89) |

|
| 2 |

|

|
99 (>95) dr = 55:45 |
— |
| 3c |

|

|
55 (64) |

|
| 4 |
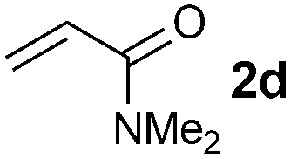
|

|
90 (87) |

|
| 5c |

|
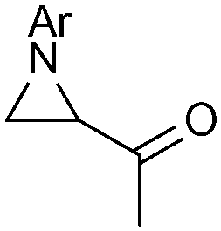
|
86 (93) | — |
| 6d |
|
|
51 (63) | Decomposition |
| 7ce |
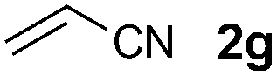
|

|
90 (>95) | — |
| 8 |
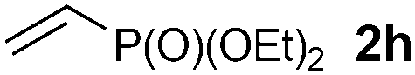
|

|
(28) | Decomposition and 1b recovered (34%) |
| 9ce |

|
|
60 (63) |

|
| 10ce |

|

|
72 (73) |
|
| 11cf |

|

|
70 (76) | Decomposition |
| 12e |
|
|
49 (57) |

|
| 13ce |

|
|
39 (40) | Decomposition |
| 14e |

|

|
36 (61) |

|
| 15g |

|

|
68 (69) |

|

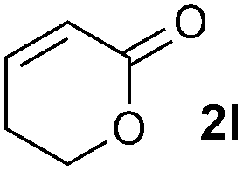
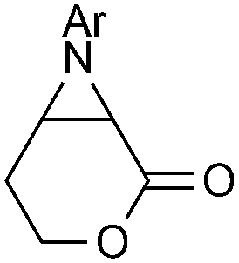
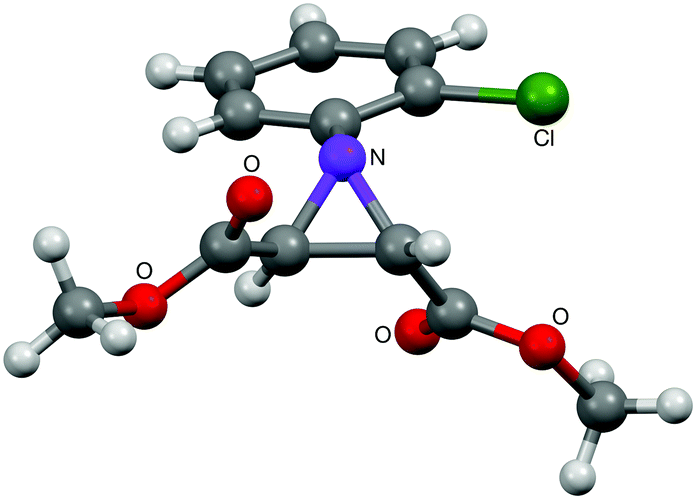
Disubstituted alkenes required higher reaction temperatures (90–110 °C) but both geminal and vicinal substitution patterns were possible (Table 2, entries 9–15). As expected, the reaction was completely stereospecific with (E)-3-penten-2-one 2n and dimethyl fumarate 2o (Table 2, entries 14 and 15) as confirmed by 1H NMR and X-ray diffraction (Fig. 1).17 In contrast, the reaction of 1b and dimethyl maleate 2p at 100 °C led to the formation of at least five different products, including both cis- and trans-4bp.18 The starting maleate was geometrically pure, so most probably the originally formed cis-4bp partially interconverted into its trans isomer at the reaction temperature.19 Next, we explored the azide scope and pleasingly these reaction conditions do not require an ortho-substituted aryl azide, which indicates that steric factors are not dominant when determining the fate of the 1,4-disubstituted triazoline. With N,N-dimethyl acrylamide 2d as model alkene, differently substituted aryl and heteroaryl azides were successfully converted into aziridines 4 (Table 3). Both electron-withdrawing and electron-donating groups led to good results with aryl azides but interestingly, very different reaction outcomes were obtained with pyridine derivatives (Table 3, entries 12 and 13). While aziridine 4md was obtained in good yield from 3-pyridylazide 1m, only low conversion into 4-pyridylaziridine 4nd was observed as the corresponding pyrazoline 6nd was obtained as the major reaction product instead.
|
|
||||
|---|---|---|---|---|
| Entry | Azide | 4Xd (%) | 7Xd (%) | |
| a Reaction conditions: Azide 1X (2 mmol), alkene 2d (4 mmol) in 4 mL of technical toluene. b Isolated yields. 1H NMR yields are given in brackets and were calculated using 1,3,5-trimethoxybenzene as internal standard and are the average of at least two independent experiments. c 10 mmol of azide 1h. d 48% (58%) of pyrazoline 6nd was obtained in this reaction. | ||||
| 1 |

|
1b | 90 (87) | 7 (9) |
| 2 |

|
1c | 89 (88) | (6) |
| 3 |

|
1d | 95 (92) | (4) |
| 4 |
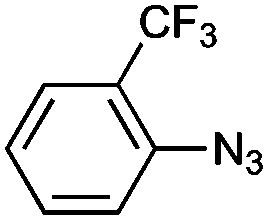
|
1e | 66 (74) | 10 (14) |
| 5 |

|
1f | 70 (82) | (4) |
| 6 |

|
1g | 90 (86) | — |
| 7c |

|
1h | 88 (>95) | — |
| 8 |
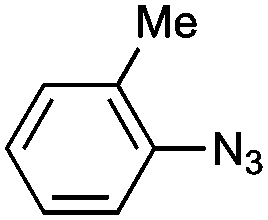
|
1i | 74 (91) | (4) |
| 9 |

|
1j | 71 (84) | (<5) |
| 10 |

|
1k | 94 (95) | (<5) |
| 11 |

|
1l | 98 (>95) | — |
| 12 |

|
1m | 83 (n.d.) | (8) |
| 13d |

|
1n | (19) | (8) |

In order to establish the synthetic practicality of these reactions, different aryl aziridines were prepared from 6.5 mmol, or more, of the starting azide 1 and several of the corresponding aziridines were obtained in the gram-scale, such as 4ba (1.15 g), 4be (1.09 g), 4bg (1.16 g), 4bo (1.20 g) or 4hd (1.94 g) (see Tables 2 and 3). Also, we adapted several protocols reported in the literature to exploit well-established reactivity of aziridines, namely nucleophilic ring-opening reactions as well as the generation of larger heterocycles. These transformations have been largely developed with activated aziridines bearing strongly electron-withdrawing groups on the nitrogen atom,20 possibly due to the lack of accessible methods for the preparation of N-alkyl- or N-aryl aziridines, often termed non-activated aziridines. Hence, we purposely used aziridine 4hd, bearing an electron rich N-aryl group to test the reactivity with three different nucleophiles, thiophenol,21 aniline,22 and trimethylsilylazide23 while avoiding any additional harsh additives (Table 4). In all cases the best results were obtained at room temperature in the absence of solvent to obtain a linear product, issue of a nucleophilic attack on the more hindered side of the starting aziridine, as the major reaction product. Excellent yields were obtained in the reaction with thiophenol and a smaller excess of nucleophile led to a slight improvement in regioselectivity (Table 4, entries 1 and 2). Compounds 8–9B were formed in good yields, but only in the presence of silica gel basified with 2% w/w aniline (Table 4, entry 3). No products were formed with either standard or NEt3-basified silica. As expected, reactions with TMSN3, the weakest nucleophile of the three, resulted in moderate conversions into 8–9C either in refluxing acetonitrile or at room temperature under neat conditions (Table 4, entries 4 and 5). Larger nucleophile excess, or increased reaction temperatures did not improve the overall results, but in all these reactions the unreacted aziridine could be recovered and no significant decomposition was observed.
|
|
||||
|---|---|---|---|---|
| Entry | Conditions | Yield (%)a |
8:9 |
|
| a Isolated yields. 1H NMR yields are given in brackets and were calculated using 1,3,5-trimethoxybenzene as internal standard and are the average of at least two independent experiments. b 42% (50%) of unreacted 4hd was also recovered. | ||||
| 1 | PhSH (7 equiv), neat, RT, 18 h | A | 99 (>95) | 80:20 |
| 2 | PhSH (2 equiv), neat, RT, 18 h | (>95) | 91:9 |
|
| 3 | PhNH2 (1.2 equiv), bas. silica gel, neat, RT, 48 h | B | 75 (84) | 80:20 |
| 4 | TMSN3 (2 equiv), MeCN, reflux, 18 h | C | 37 (46)b | 82:12 |
| 5 | TMSN3 (4 equiv), neat, RT, 18 h | 51 (63) | 94:6 |
|

Next, we used this methodology as starting point to easily access larger cyclic molecules. The synthesis of pyrrolidine 10 from 4ha and N-phenylmaleimide in anhydrous and degassed refluxing toluene had been previously reported in 48% yield.24 Gratifyingly, both cycloaddition steps could be carried out in one pot in technical toluene and in the presence of air, by simply adding the maleimide and raising the temperature after 16 h to generate the corresponding azomethine ylide. Pyrrolidine 10 was then isolated in an overall 71% yield for both steps (Scheme 3A).25 Finally, 2-oxazolidone 11 was prepared by reacting aziridine 4hd with methyl chloroformate, conditions previously reported for N-benzyl aziridines.26 In this case different solvents had to be used for each step as the one-pot reaction in toluene led to the formation of an uncyclised α-chlorocarboxylate intermediate as final product.18 Switching to refluxing acetonitrile for the reaction with chloroformate cleanly led to oxazolidone 11 in an excellent yield (Scheme 3B).
In conclusion, the present methodology is convenient, user friendly, and easily scalable. It does not require a metal catalyst or additional reagents such as strong Brønsted acids. Furthermore, it offers a complementary approach to other established reactions since it does not require an electron-poor nitrogen source and it allows the introduction of a range of polar functional groups, which makes the resulting aziridines particularly useful as building blocks.27 Except for highly activated alkenes, the reaction remains stereospecific and the geometry of the starting alkene is preserved in the final heterocycle. The sheer simplicity of these reactions together with the known asymmetric transformations of racemic aziridines28 makes them an attractive alternative for the broader synthetic community.
 | ||
| Scheme 3 Ring-expansion reactions of aryl aziridines. | ||
EPSRC and the Royal Thai Government are acknowledged for studentships to F. S. and L. R., respectively.
Conflicts of interest
There are no conflicts to declare.Notes and references
- Aziridines and Epoxides in Organic Synthesis, ed., A. K. Yudin, Wiley-VCH, 2006 Search PubMed.
- (a) L. Degennaro, P. Trinchera and R. Luisi, Chem. Rev., 2014, 114, 7881–7929 CrossRef CAS PubMed; (b) H. Pellissier, Tetrahedron, 2010, 66, 1509–15555 CrossRef CAS; (c) P. Muller and C. Fruit, Chem. Rev., 2003, 103, 2905–2919 CrossRef PubMed.
- E. Roma, E. Tosi, M. Miceli and T. Gasperi, Asian J. Org. Chem., 2018, 7, 2357–2367 CrossRef CAS.
- A. Yoshimura, K. R. Middleton, C. Zhu, V. N. Nemykin and V. V. Zhdankin, Angew. Chem., Int. Ed., 2012, 51, 8059–8062 CrossRef CAS PubMed ; See also: (a) J. Li, P. W. H. Chan and C.-M. Che, Org. Lett., 2005, 7, 5801–5804 CrossRef CAS PubMed; (b) L. B. Krasnova and A. K. Yudin, Org. Lett., 2006, 8, 2011–2014 CrossRef CAS PubMed.
- J. J. Farndon, T. A. Young and J. F. Bower, J. Am. Chem. Soc., 2018, 140, 17846–17850 CrossRef CAS PubMed.
- (a) J. Li, W. Huang, J. Chen, L. He, X. Cheng and G. Li, Angew. Chem., Int. Ed., 2018, 57, 5695–5698 CrossRef CAS PubMed; (b) D. E. Holst, D. J. Wang, M. J. Kim, I. A. Guzei and Z. K. Wickens, Nature, 2021, 596, 74–79 CrossRef CAS PubMed ; See also: ; (c) T. Siu and A. K. Yudin, J. Am. Chem. Soc., 2002, 124, 530–531 CrossRef CAS PubMed; (d) J. Chen, W.-Q. Yan, C. M. Lam, C.-C. Zeng, L.-M. Hu and R. D. Little, Org. Lett., 2015, 17, 986–989 CrossRef CAS PubMed.
- J. Vesely, I. Ibrahem, G.-L. Zhao, R. Rios and A. Córdova, Angew. Chem., Int. Ed., 2007, 46, 778–781 CrossRef CAS PubMed . See also: A. Armstrong, C. A. Baxter, S. G. Lamont, A. R. Pape and R. Wincewicz, Org. Lett., 2007, 9, 351–353 CrossRef PubMed.
- N. Jung and S. Bräse, Angew. Chem., Int. Ed., 2012, 51, 5538–5540 CrossRef CAS PubMed.
- R. Huisgen, in 1,3-Dipolar Cycloaddition Chemistry, ed. A. Padwa, Wiley-Interscience, New York, 1984, vol. 1, pp. 1–176 Search PubMed.
- (a) R. Huisgen, G. Szeimies and L. Möbius, Chem. Ber., 1966, 99, 475–490 CrossRef CAS; (b) G. Szeimies and R. Huisgen, Chem. Ber., 1966, 99, 491–503 CrossRef CAS.
- (a) W. Broeckx, N. Overbergh, C. Samyn, G. Smets and G. L’Abbé, Tetrahedron, 1971, 27, 3527–3534 CrossRef CAS; (b) M. Chen, Y. Gan and L. M. Harwood, Synlett, 2008, 2119–2121 CAS; (c) D. M. Reddy, N. A. Qazy, S. D. Sawant, A. H. Bandey, J. Srinivas, M. Shankar, S. K. Singh, M. Verma, G. Chashoo, A. Saxena, D. Mondhe, A. K. Saxena, V. K. Sethi, S. C. Taneja, G. N. Qazi and H. M. S. Kumar, Eur. J. Med. Chem., 2011, 46, 3210–3217 CrossRef CAS PubMed; (d) S. J. Gharpure, S. Naveen, G. Samala and D. S. Vishwakarma, Chem. – Eur. J., 2019, 25, 1456–1460 CrossRef CAS PubMed.
- (a) S. Allemann and P. Vogel, Synthesis, 1991, 923–928 CrossRef CAS; (b) R. S. Dahl and N. S. Finney, J. Am. Chem. Soc., 2002, 126, 8356–8357 CrossRef PubMed; (c) T. S. Chung, S. A. Lopez, K. N. Houk and M. A. Garcia-Garibay, Org. Lett., 2015, 17, 4568–4571 CrossRef CAS PubMed.
- J. Bourgois, M. Bourgois and F. Texier, Bull. Soc. Chim. Fr., 1978, 9–10, 485–527 Search PubMed.
- (a) J. M. Mahoney, C. R. Smith and J. N. Johnston, J. Am. Chem. Soc., 2005, 127, 1354–1355 CrossRef CAS PubMed; (b) J. Li, Y. Fu and Q.-X. Guo, Tetrahedron, 2008, 64, 11167–11174 CrossRef CAS.
- F. Sebest, K. Lachhani., C. Pimpasri, L. Casarrubios, A. J. P. White, H. S. Rzepa and S. Díez-González, Adv. Synt. Catal., 2020, 362, 1877–1886 CrossRef CAS.
- C. Capello, U. Fischer and K. Hungerbühler, Green Chem., 2007, 9, 927–934 RSC.
- (a) P. Scheiner, J. Am. Chem. Soc., 1966, 88, 4759–4760 CrossRef CAS; (b) F. Sebest, L. Casarrubios, H. S. Rzepa, A. J. P. White and S. Díez-González, Green Chem., 2018, 20, 4023–4035 RSC.
- See ESI† for further details.
- R. Huisgen, W. Scheer and H. Huber, J. Am. Chem. Soc., 1967, 89, 1753–1755 CrossRef CAS.
- G. S. Singh, Adv. Heterocycl. Chem., 2018, 129, 245–335 CrossRef.
- Y. Zhou and P. V. Murphy, Org. Lett., 2008, 10, 3777–3780 CrossRef CAS PubMed.
- R. V. Anand, G. Pandey and V. K. Singh, Tetrahedron Lett., 2002, 43, 3975–3976 CrossRef CAS.
- M. Chandrasekhar, G. Sekar and V. K. Singh, Tetrahedron Lett., 2000, 41, 10079–10083 CrossRef CAS.
- P. DeShong, D. A. Kell and D. R. Sidler, J. Org. Chem., 1985, 50, 2309–2315 CrossRef CAS.
- The procedure reported in reference 24 requires 14 days in the dark and two sublimation steps to isolate aziridine 4ha in less than 50% yield. Aziridine 4ha could be isolated in 90% yield using our standard methodology, see ESI† for full details.
- T. B. Sim, S. H. Kang, K. S. Lee, W. K. Lee, H. Yun, Y. Dong and H. J. Ha, J. Org. Chem., 2003, 68, 104–108 CrossRef CAS PubMed.
- A. S. Pankova and M. A. Kuznetsov, Synthesis, 2017, 5093–5104 CAS.
- Z. Chai, Synthesis, 2020, 1738–1750 CrossRef CAS.
- F. Sebest, L. Radtanajiravong, S. Kaukver, A. J. P. White and S. Díez-González, Imperial College Research Services Data Repository, 2021 DOI:10.14469/hpc/9032 and sub-collections therein.
Footnotes |
| † Electronic supplementary information (ESI) available: FAIR data for NMR spectra and crystallographic data, see ref. 29. CCDC 2129406. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1cc07213g |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2022 |