
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Operando transmission electron microscopy of battery cycling: thickness dependent breaking of TiO2 coating on Si/SiO2 nanoparticles†
Shibabrata
Basak
 *abc,
Amir H.
Tavabi
b,
Krzysztof
Dzieciol
a,
Vadim
Migunov
b,
Violetta
Arszelewska
d,
Hermann
Tempel
a,
Hans
Kungl
a,
Erik M.
Kelder
d,
Marnix
Wagemaker
d,
Chandramohan
George
c,
Joachim
Mayer
be,
Rafal E.
Dunin-Borkowski
b and
Rüdiger-A.
Eichel
af
*abc,
Amir H.
Tavabi
b,
Krzysztof
Dzieciol
a,
Vadim
Migunov
b,
Violetta
Arszelewska
d,
Hermann
Tempel
a,
Hans
Kungl
a,
Erik M.
Kelder
d,
Marnix
Wagemaker
d,
Chandramohan
George
c,
Joachim
Mayer
be,
Rafal E.
Dunin-Borkowski
b and
Rüdiger-A.
Eichel
af
aInstitute of Energy and Climate Research, Fundamental Electrochemistry (IEK-9), Forschungszentrum Jülich GmbH, 52425 Jülich, Germany. E-mail: s.basak@fz-juelich.de
bErnst Ruska-Centre for Microscopy and Spectroscopy with Electrons and Peter Grünberg Institute, Forschungszentrum Jülich GmbH, 52425 Jülich, Germany
cDyson School of Design Engineering, Imperial College London, SW7 2AZ, London, UK
dDepartment of Radiation Science and Technology, Delft University of Technology, Mekelweg 15, Delft, 2629JB, The Netherlands
eCentral Facility for Electron Microscopy (GFE), RWTH Aachen University, 52074 Aachen, Germany
fInstitute of Physical Chemistry, RWTH Aachen University, 52074 Aachen, Germany
First published on 31st January 2022
Abstract
Conformal coating of silicon (Si) anode particles is a common strategy for improving their mechanical integrity, to mitigate battery capacity fading due to particle volume expansion, which can result in particle crumbling due to lithiation induced strain and excessive solid–electrolyte interface formation. Here, we use operando transmission electron microscopy in an open cell to show that TiO2 coatings on Si/SiO2 particles undergo thickness dependent rupture on battery cycling where thicker coatings crumble more readily than thinner (∼5 nm) coatings, which corroborates the difference in their capacities.
Si has gained considerable traction as an anode material in Li-ion batteries, in which composite electrodes that have different amounts of Si and carbon1–3 (with a theoretical capacity for Si of above ∼4000 mA h g−1 based on a Li22S5 stoichiometry) offer an enhancement in capacity when compared with pure graphite powder (∼370 mA h g−1). However, the breaking of Si particles because of strain caused by repeated lithiation (during battery charge/discharge cycles) limits exploitation of their full capacity and rate capabilities. In particular, the large volume change (>300%) of Si particles4,5 upon lithiation can result in particle pulverisation,6 accompanied by excessive solid electrolyte interphase (SEI) formation. The size-dependent cracking7 and shape-dependent lithiation8 behavior of Si has been reported, which provided indications about how the mechanical characteristics of Si particles evolve during battery cycling and affect their electrochemical performance. Even nanosized particles that do not easily break during charge/discharge cycles owing to improved strain relaxation are still plagued by excessive electrolyte consumption associated with multiple SEI formation events, which causes rapid depletion of cyclable Li. The surface coating of Si-based nanostructures offers a tradeoff between mechanical integrity and electrochemical performance. Among a wide range of surface coatings for Si/SiO2, including Cu,9 C,10,11 polymers,12 alumina13 and carbides,14 the coating of Si particles with titanium oxide (TiO2)15,16 is highly appealing as TiO2 is converted to lithium titanate on lithiation via the reaction TiO2 + Li+ + e− ↔ LiTiO2, offering not only high rate capability but also long-term stability during repeated lithiation/delithiation cycles by preserving active particle cores and reducing SEI formation, thereby mitigating battery capacity decay and improving battery longevity. The use of a metal oxide coating on Si also makes charge/discharge processes less exothermic17 when compared to pure Si, contributing to battery safety. Despite the beneficial role of such coatings18,19 for protecting active particles from parasitic reactions (electrolyte decomposition) and particle pulverization (via strain dissipation), their role in mediating reactions with Li during repeated lithiation/delithiation is not yet understood. Postmortem (ex situ) electrode analysis has been used to infer that coated active particles exhibited minimal particle cracking/fracture, in alignment with their better electrochemical performance when compared with their uncoated counterparts.
The development of a local understanding of the behavior of such coatings during lithiation/delithiation requires direct visualization of individual particles during cycling via in situ transmission electron microscopy (TEM). Liquid cell in situ TEM has been used to study Li dendrites on gold-based electrodes,20 mechanistic pathways associated with MoS2 lithiation,21 and sodium metal nucleation22 on electrode with different surfaces. Solid-state cell in situ TEM has provided insight about Si/polypyrrole23 anodes, for which a polypyrrole coating on Si improves mechanical properties. However, studies involving metal oxides (e.g., TiO2) that are the most common coating materials are still limited. The thicknesses of such coatings are an important consideration because they can affect both the mechanical interaction (between coating and active particles) and mass loading of active particles. Therefore, it is important to assess how coating thickness can influence battery capacity upon cycling. Here, we perform an operando TEM study of the lithiation/delithiation of Si/SiO2 nanoparticles (average core diameter ∼100 nm) coated with two different TiO2 coating thicknesses (∼5 and ∼10 nm) using a solid-state electrolyte (LixO).
We previously showed that Si nanoparticles24 that are not in direct contact with solid–electrolyte (in operando) can still be lithiated over a few tens to hundreds of nanometers, as the lithiation reaction is mediated by interparticle connectivity. Here, we coat similar Si nanoparticles with two different thicknesses of TiO2 using atomic layer deposition (ALD) in a fluidized bed reactor at 150 °C. Titanium isopropoxide with water was used as the co-reactant, with the coating thicknesses depending on the number of coating cycles. Details about the coating procedure can be found in the ESI.† A thin native Si oxide layer, which passivates the Si nanoparticles, can be observed in Fig. 1a. Fig. 1b and c show energy dispersive X-ray spectroscopy mapping of Si nanoparticles for TiO2 coating thicknesses of ∼5 and ∼10 nm, respectively. The TiO2 coating envelops the Si nanoparticles, either individually or as clusters, around the native oxide layer. These nanoparticles were used to assemble batteries in a Nanofactory TEM specimen holder, with Li/LixO on a tungsten (W) needle serving as a solid-state electrolyte in an open cell configuration, as shown in Fig. 2a and in bright field TEM of Fig. 2b. Details about the battery assembly can be found in the ESI.† High resolution TEM and scanning TEM images of bare Si/SiO2 particles as well as TiO2 coated Si/SiO2 particles are shown in Fig. S1 of the ESI.†Fig. 3 shows Si particles coated with 5 nm thick TiO2, which were largely preserved upon lithiation (Fig. 3a–h) and delithiation (Fig. 3i–p). Despite seldom coatings rupture on these nanoparticles, they were mostly observed to be intact. Complete lithiation/delithiation cycles of two different 5 nm TiO2 coated Si nanoparticle clusters can be followed in Movies S1 and S2 in the ESI† (both movies plays at 24× speed). It should be noted that the coated Si nanoparticles are also likely to rely on interparticle connectivity for (de)lithiation, because many nanoparticles that are not directly in contact with the electrolyte (LixO) are able to expand in size (lithiation).
 | ||
| Fig. 1 Energy dispersive X-ray spectroscopy mapping of Si nanoparticles used in operando experiments. (a) Si nanoparticles with an oxide layer, (b) Si nanoparticles coated with ∼5 nm of TiO2, and (c) Si nanoparticles coated with ∼10 nm of TiO2. | ||
 | ||
| Fig. 2 (a) Schematics of transmission electron microcopy (TEM) half copper grid with titanium oxide (TiO2) coated silicon/silicon oxide (Si/SiO2) nanoparticles and tungsten needle scratched against Li metal, which contains a Li/LixO fragment as a solid-state electrolyte. (b) Bright-field TEM image showing the open cell configuration used to perform lithiation/delithiation of Si/SiO2 nanoparticles coated with TiO2. | ||
 | ||
| Fig. 3 Bright-field transmission electron microcopy (TEM) images recorded at different stages of the lithiation/delithiation of silicon (Si/SiO2) nanoparticles coated with ∼5 nm of titanium oxide. (a–h) lithiation and (i–p) subsequent delithiation. Scale bar is 100 nm. | ||
In contrast, clusters of Si nanoparticles with a thicker (∼10 nm) TiO2 coating have a greater tendency to rupture. Fig. 4(a–h) and (i–p) show different stages of lithiation and delithiation, respectively. The complete (de)lithiation process of the 10 nm TiO2 coated particles can be seen in Movie S3 in the ESI.† Movie S4 (ESI†) shows electrochemical lithiation of another Si nanoparticle cluster with 10 nm TiO2 coated particles (both movies plays at 24× speed). By comparing the responses of Si nanoparticles with the two different coating thicknesses, the use of a ∼10 nm TiO2 coating can be seen to result in cracks and breakage compared with a ∼5 nm TiO2 coating. Up to 15% of the 10 nm TiO2 coated particles were observed to break during in situ battery cycling. All experiments were performed at <10 e Å−2 s−1 dose rate, which is similar to that used for biological samples, to ensure the electron beam does not influence the observation. Given that the same sizes of Si nanoparticles, similar amorphous coatings (Fig. S1 and S4 of ESI†) and identical cycling conditions were used for both cases, the coating rupture appears to be influenced primarily by the coating thickness. It should be noted that cracks that had formed during lithiation cycles in the ∼10 nm TiO2 coated particles were able to restore by reconnecting to some extent (see the Movie S3 in the ESI†). However, this (all solid-state) observation may not be reproduced in cells that have liquid electrolytes, in which direct contact between Si and the liquid electrolyte could occur, leading to new SEI formation, preventing cracks from reconnecting during further cycles.
 | ||
| Fig. 4 Bright-field transmission electron microcopy (TEM) images recorded at different stages of the lithiation/delithiation of silicon (Si/SiO2) nanoparticles coated with ∼10 nm of titanium oxide. (a–h) Lithiation and (i–p) subsequent delithiation. Scale bar is 100 nm. | ||
Therefore, we compared the electrochemical performance of electrodes made with uncoated Si, 5 nm TiO2 coated Si and 10 nm TiO2 coated Si nanoparticles in Li half-cells using a liquid electrolyte. Details of electrode preparation and battery assembly can be found in the ESI.†Fig. 5 shows that all three electrodes show almost similar capacities at the first cycle at C/10 (∼1500 mA h g−1), but markedly different rate capabilities. Si nanoparticles with a thinner (∼5 nm) TiO2 coating delivered capacities up to 1000 mA h g−1 during subsequent cycles at C/10, while particles with a thicker (∼10 nm) TiO2 coating showed capacities below 500 mA h g−1 at the same rates, indicating that Si nanoparticles with 5 nm TiO2 coating have better capacity as well as cycling stability, while Si nanoparticles with 10 nm coating exhibit a gradual decrease in capacity. Interestingly, the uncoated Si nanoparticles also have a capacity of ∼500 mA h g−1 at C/10, which is more stable than Si nanoparticles with the 10 nm TiO2 coating. A similar trend is observed at the C/5 and C/2 rates. At C/2, the electrode with Si nanoparticles with the thinner (∼5 nm) TiO2 coating offers stable capacities up to ∼750 mA h g−1, while the capacity of uncoated Si nanoparticles appears stable but is lower at ∼400 mA h g−1. In contrast, electrodes with Si nanoparticles with a thicker (∼10 nm) TiO2 coating exhibit the lowest capacity (∼250 mA h g−1), as well as rapid capacity fading. Differences in capacities (from cells with liquid electrolytes) can be rationalized based on the operando TEM observations (solid-state). During the first cycle, electrochemical lithiation of the TiO2 coating, (de)lithiation of Si and SEI formation take place. The electrode with the ∼10 nm TiO2 coated nanoparticles showed the highest capacity, likely due to lithiation of excess TiO2, followed by the ∼5 nm TiO2 coated nanoparticles and the uncoated nanoparticles in terms of capacity values. However, starting from the second cycle the battery is cycled between 1.2 and 0.01 V vs. Li/Li+, the TiO2 coating is already lithiated and it does not participate actively in further electrochemical lithiation reactions. The fact that only (de)lithiation of Si and SEI formation take place from the second cycle can be inferred from the observation that the electrode with the ∼5 nm TiO2 coated nanoparticles shows a higher capacity of ∼1000 mA h g−1, while both the ∼10 nm TiO2 coated nanoparticles and the uncoated nanoparticles show much lower capacities (∼500 mA h g−1).
 | ||
| Fig. 5 Electrochemical performance of different silicon anodes (Si/SiO2 with no coating, with 5 nm TiO2 coating and with 10 nm TiO2 coating) at different C rates. The first 5 cycles are at C/10, at C/5 and at C/2 rates. | ||
Such a large difference in capacity loss from the first cycle can be attributed to excessive SEI formation in the case of the ∼10 nm TiO2 coated nanoparticles and the uncoated nanoparticles. In principle, the capacity of the ∼10 nm TiO2 coated nanoparticles should not be affected by SEI formation to a large extent because the active nanoparticles are coated by TiO2, which should protect the silicon surfacefrom electrolyte exposure. However, the in situ TEM experiments (solid-state) show that the nanoparticles start to crumble even during the first cycle and the coatings break readily, providing access to Si–liquid electrolyte interaction in a liquid electrolyte environment (Fig. 5) and leading to excess SEI formation. Repeated SEI formation occurs as new surfaces are created because of particle breaking, resulting in capacity degradation. For the ∼5 nm TiO2 coated nanoparticles that undergo almost no/seldom breaking, no further capacity loss due to excess SEI formation is observed, as depicted schematically (for all the three types of particles tested) in Fig. 5. We consider two possible scenarios to explain our observations. In the first scenario, the ∼5 nm TiO2 coated nanoparticles, despite occasional initial cracks, having a thinner coating that attaches to the nanoparticles more strongly and protects them from direct contact in a liquid electrolyte, thus mitigating continuous SEI formation. This inference is consistent with the measured capacity retention (Fig. 5), which follows the order: 5 nm TiO2 coated Si nanoparticles > uncoated Si nanoparticles > 10 nm TiO2 coated Si nanoparticles. In the second scenario, the improved resistance of the thinner coating to breakage results from its higher fracture resistance and strain tolerance. To gain more insight into the swelling behavior of the 5 nm and 10 nm TiO2 coated Si nanoparticles, their first lithiation and the corresponding expansion in size is plotted as a function of time, as in Fig. 6. Note that, as the particle's boundaries are not well defined, area fraction calculation was performed based on manual segmentation of the acquired images. The gradual swelling of the 5 nm TiO2 coated particles for area calculation is shown in Fig. S2 (ESI†).
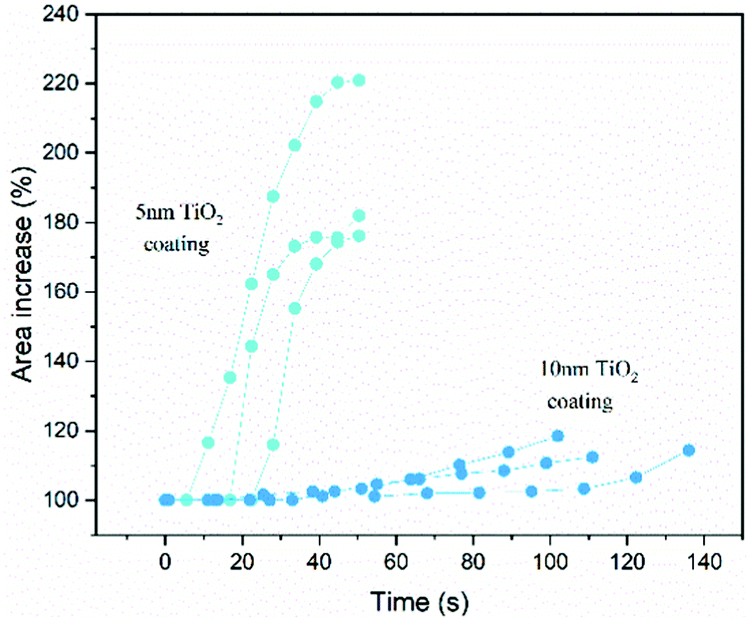 | ||
| Fig. 6 Changes in the areas of different nanoparticles plotted as a function of time during lithiation. | ||
Ex situ TEM analysis has been reported that for 100 nm particles coated with 3 nm of amorphous TiO2,15 the coating protects the particles more efficiently from crumbling owing to better strain relaxation than a more crystalline coating. Similarly, ex situ microscopic analysis of micron-sized Si with different thicknesses of SiO225 has been reported that an optimal coating thickness of ∼7 nm facilitated stable capacity retention, whereas increased thickness led to poorer electrochemical performance and decreased CE%. In our in situ TEM study, Fig. 6 shows that ∼5 nm TiO2 coated nanoparticles remain intact even after ≥180% expansion, while ∼10 nm TiO2 coated nanoparticles break within ≤120% expansion, suggesting that the ∼10 nm TiO2 coated nanoparticles are more susceptible to breakage even with a lesser degree of lithiation. With seldom rupture, the ∼5 nm TiO2 coated nanoparticles were able to expand to ≥180%, implying the breakage of thicker coating is due to increased stress compared with thinner coating. This in turn determines the extent to which Si surface is exposed to electrolyte (in liquid cells) and electrolyte consumption (via SEI) as more particles break upon cycling, resulting in different battery capacities (as in Fig. 5), thus revealing the crucial role of coating thickness.
In conclusion, our operando TEM of battery cycling in an open cell showed that thicker TiO2 coatings on Si/SiO2 nanoparticles tend to break more readily compared to thinner coatings, corroborating their cycling behavior and capacity measured using cells with liquid electrolytes. Our operando TEM studies contribute to the design of metal oxide coatings, which are required for improved cycling stability and battery capacity.
SB acknowledges Marie Skłodowska-Curie fellowship ‘Electroscopy’ (Grant no. 892916). EMK acknowledges financial support from Shell Global Solutions International BV through a contracted research agreement. CG acknowledges funding from the Royal Society, London for a URF (Grant no. UF160573).
Conflicts of interest
There are no conflicts to declare.References
- S. Chae, N. Kim, J. Ma, J. Cho and M. Ko, Adv. Energy Mater., 2017, 7(15), 1700071 CrossRef.
- F. Dou, L. Shi, G. Chen and D. Zhang, Electrochem. Energy Rev., 2019, 2(1), 149–198 CrossRef CAS.
- Y. Jin, B. Zhu, Z. Lu, N. Liu and J. Zhu, Adv. Energy Mater., 2017, 7(23), 1700715 CrossRef.
- M. T. McDowell, S. W. Lee, W. D. Nix and Y. Cui, Adv. Mater., 2013, 25(36), 4966–4985 CrossRef CAS PubMed.
- M. Ashuri, Q. He and L. L. Shaw, Nanoscale, 2016, 8(1), 74–103 RSC.
- M. T. McDowell, I. Ryu, S. W. Lee, C. Wang, W. D. Nix and Y. Cui, Adv. Mater., 2012, 24(45), 6034–6041 CrossRef CAS PubMed.
- X. H. Liu, L. Zhong, S. Huang, S. X. Mao, T. Zhu and J. Y. Huang, ACS Nano, 2012, 6(2), 1522–1531 CrossRef CAS PubMed.
- C. K. Chan, H. Peng, G. Liu, K. McIlwrath, X. F. Zhang, R. A. Huggins and Y. Cui, Nat. Nanotechnol., 2008, 3(1), 31–35 CrossRef CAS PubMed.
- S. Murugesan, J. T. Harris, B. A. Korgel and K. J. Stevenson, Chem. Mater., 2012, 24(7), 1306–1315 CrossRef CAS.
- Y.-S. Hu, R. Demir-Cakan, M.-M. Titirici, J.-O. Müller, R. Schlögl, M. Antonietti and J. Maier, Angew. Chem., Int. Ed., 2008, 47(9), 1645–1649 CrossRef CAS PubMed.
- D. J. Lee, M.-H. Ryou, J.-N. Lee, B. G. Kim, Y. M. Lee, H.-W. Kim, B.-S. Kong, J.-K. Park and J. W. Choi, Electrochem. Commun., 2013, 34, 98–101 CrossRef CAS.
- H. Wu, G. Yu, L. Pan, N. Liu, M. T. McDowell, Z. Bao and Y. Cui, Nat. Commun., 2013, 4(1), 1943 CrossRef PubMed.
- H. T. Nguyen, M. R. Zamfir, L. D. Duong, Y. H. Lee, P. Bondavalli and D. Pribat, J. Mater. Chem., 2012, 22(47), 24618–24626 RSC.
- C. Yu, X. Chen, Z. Xiao, C. Lei, C. Zhang, X. Lin, B. Shen, R. Zhang and F. Wei, Nano Lett., 2019, 19(8), 5124–5132 CrossRef CAS PubMed.
- J. Yang, Y. Wang, W. Li, L. Wang, Y. Fan, W. Jiang, W. Luo, Y. Wang, B. Kong, C. Selomulya, H. K. Liu, S. X. Dou and D. Zhao, Adv. Mater., 2017, 29(48), 1700523 CrossRef PubMed.
- J. John, B. Gangaja, S. V. Nair and D. Santhanagopalan, Electrochim. Acta, 2017, 235, 191–199 CrossRef CAS.
- G. Jeong, J.-H. Kim, Y.-U. Kim and Y.-J. Kim, J. Mater. Chem., 2012, 22(16), 7999–8004 RSC.
- T. Song and U. Paik, J. Mater. Chem. A, 2016, 4(1), 14–31 RSC.
- A. Gao, S. Mukherjee, I. Srivastava, M. Daly and C. V. Singh, Adv. Mater. Interfaces, 2017, 4(23), 1700920 CrossRef.
- Z. Zeng, W.-I. Liang, H.-G. Liao, H. L. Xin, Y.-H. Chu and H. Zheng, Nano Lett., 2014, 14(4), 1745–1750 CrossRef CAS PubMed.
- Z. Zeng, X. Zhang, K. Bustillo, K. Niu, C. Gammer, J. Xu and H. Zheng, Nano Lett., 2015, 15(8), 5214–5220 CrossRef CAS PubMed.
- Z. Zeng, P. Barai, S.-Y. Lee, J. Yang, X. Zhang, W. Zheng, Y.-S. Liu, K. C. Bustillo, P. Ercius, J. Guo, Y. Cui, V. Srinivasan and H. Zheng, Nano Energy, 2020, 72, 104721 CrossRef CAS.
- L. Luo, P. Zhao, H. Yang, B. Liu, J.-G. Zhang, Y. Cui, G. Yu, S. Zhang and C.-M. Wang, Nano Lett., 2015, 15(10), 7016–7022 CrossRef CAS PubMed.
- S. Basak, V. Migunov, A. H. Tavabi, C. George, Q. Lee, P. Rosi, V. Arszelewska, S. Ganapathy, A. Vijay, F. Ooms, R. Schierholz, H. Tempel, H. Kungl, J. Mayer, R. E. Dunin-Borkowski, R.-A. Eichel, M. Wagemaker and E. M. Kelder, ACS Appl. Energy Mater., 2020, 3(6), 5101–5106 CrossRef CAS.
- S. Sim, P. Oh, S. Park and J. Cho, Adv. Mater., 2013, 25(32), 4498–4503 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1cc07172f |
| This journal is © The Royal Society of Chemistry 2022 |