
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceControlling spontaneous chirality in achiral materials: liquid crystal oligomers and the heliconical twist-bend nematic phase†
M. M.
Majewska
 *a,
E.
Forsyth
b,
D.
Pociecha
a,
C.
Wang
c,
J. M. D.
Storey
b,
C. T.
Imrie
b and
E.
Gorecka
a
*a,
E.
Forsyth
b,
D.
Pociecha
a,
C.
Wang
c,
J. M. D.
Storey
b,
C. T.
Imrie
b and
E.
Gorecka
a
aFaculty of Chemistry, University of Warsaw, ul. Zwirki i Wigury 101, 02-089 Warsaw, Poland. E-mail: mmajewska@chem.uw.edu.pl
bDepartment of Chemistry, School of Natural and Computing Sciences, University of Aberdeen, Meston Building, Aberdeen AB24 3UE, UK
cAdvanced Light Source, LBNL, Berkeley, CA 94720, USA
First published on 10th April 2022
Abstract
Liquid crystal oligomers, namely dimers, trimers and tetramers, consisting of cyanobiphenyl and benzylideneaniline-based mesogenic units connected by either linear or bent alkoxy or alkyl spacers are reported. These materials, although built from achiral molecules, show the spontaneously chiral heliconical twist-bend nematic (NTB) phase. We report the relationships between the shape of the oligomer, and the NTB phase stability, the temperature dependence of the helical pitch length and tilt angle, birefringence, and elastic constants.
The spontaneous formation of chirality in a system composed of achiral molecules is of fundamental and technological importance in both the physical and biological sciences, and as such hugely topical.1 Spontaneous mirror symmetry breaking has been observed in various liquid crystalline phases,2 the first fluid to exhibit this phenomenon in the absence of positional order was the twist-bend nematic phase (NTB).3–5 More recently, spontaneous chirality has also been confirmed in an isotropic liquid.6 In the NTB phase the molecules more or less point along the same direction known as the director, and the director forms a helix and is tilted with respect to the helical axis; statistically equal numbers of the degenerate left and right-handed helices form. The pitch length is remarkably short, just a few nanometres.5 The molecular structural prerequisite for the observation of the NTB phase is an overall bent shape, and the overwhelming majority of twist-bend nematogens are odd-membered liquid crystal dimers.7 A dimer consists of two mesogenic units attached via a flexible spacer, normally an alkyl chain. The degeneracy of the helices may be lifted if the molecules are chiral and the chiral NTB phase is seen.3 More recently, twist-bend smectic phases have also been reported for achiral odd-membered dimers8,9 and these exhibit multiple levels of chirality even though their constituent molecules are achiral. An absolutely central question in our understanding of these chiral structures assembled from achiral molecules is how to control the chirality with a view to designing new materials with tailored properties such as pitch length and its temperature dependence. It has been shown that the pitch length and its temperature dependence for NTB phases exhibited by dimers and trimers are very different.10
Very few reports of higher oligomers that form the NTB phase have appeared.7,11–20 Here we report the first extensive and systematic study of oligomers with identical mesogenic units in order to better understand how the tendency to exhibit spontaneous chirality depends on molecular structure. The structures and acronyms of the oligomers are shown in Fig. 1 and Fig. S1 (ESI†). In describing these structures, and in order to highlight the essential role played by molecular shape, we use general designations, in which B refers to a linkage with an odd number of atoms which imposes a bent arrangement of neighbouring units, and L to a linkage with an even number of atoms which gives a linear arrangement of neighbouring units (Fig. S2, ESI†). The phase transition temperatures, together with associated entropy changes are given in Table S1 (ESI†) and shown in Fig. S3 (ESI†). The relative stabilities of the NTB phases for the dimer (CB6OCB), trimer (CB6O.O6CB), and tetramer (CB6O.6O.O6CB), all having the same hexyloxy flexible spacers (B), are compared in Fig. 1 in terms of a scaled transition temperature, and it was found that this increases monotonically with increasing number of mesogenic units in the oligomer. The absolute NTB–N (TNTBN) and N-Iso (TNI) transition temperatures also increase passing from the dimer to trimer to tetramer (Fig. S3, ESI†), by around 30 K and 10 K, respectively. Previous studies have focussed on the evolution of liquid crystal behaviour from a conventional low molar mass mesogen containing a single mesogenic unit to the corresponding dimer and trimer, and revealed a much smaller increase in TNI on passing from the dimer to trimer than from the monomer to dimer.21,22 The data reported here indicate that this trend continues on passing from the trimer to tetramer, and suggests that the mesogenic units in the oligomeric species are correlated to about the same extent. A comparison of the transition temperatures shown by the tetramers that differ only in the central flexible spacer reveals that increasing the molecular bend angle imposed by the central spacer (bend angle C7 < OC6 < OC5O < OC6O)23 is accompanied by an increase in TNI (Fig. S3, ESI†).
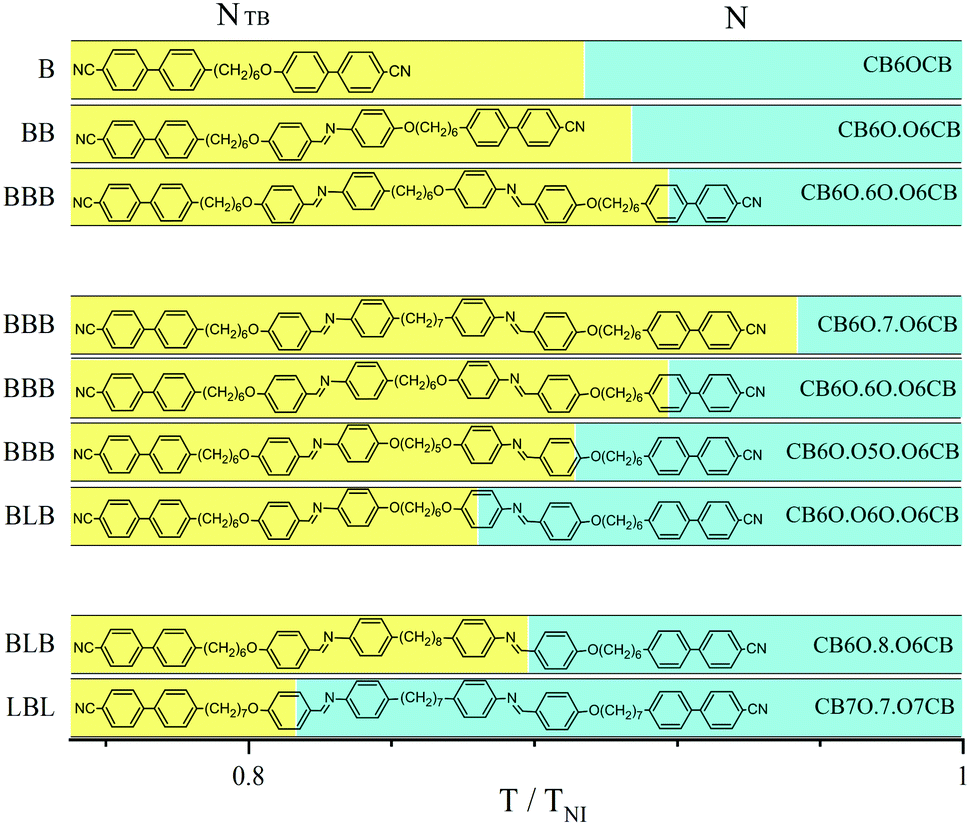 | ||
| Fig. 1 The temperature range of the NTB (yellow) and N (cyan) phases relative to TNI: for a dimer, trimer and tetramer with hexyloxy (OC6) linkages (upper part), for tetramers with different central linkages C7, OC6, OC5O, OC6O and hence different molecular bend angles (middle part), and for two tetramers having differing sequences of B (bent) and L (linear) linkages (lower part). Melting points are not shown here for the sake of clarity, see Fig. S3 (ESI†). | ||
This may be accounted for in terms of the enhanced structural anisotropy. It is noteworthy, however, that the effect on TNI of changing the central spacer in the tetramer is much smaller than making the same change in a dimer.24 This suggests for the tetramers that orientationally the four mesogenic units are not as strongly correlated via the central spacer, and that the average shapes of the tetramers depend to a greater extent on the two outer spacers. The monotonic decrease of the relative stability of the NTB phase (Fig. 1) on increasing the molecular bend angle suggests that the NTB–N transition is predominantly shape-driven. In the final comparison, two tetramers differing in the sequence of B and L linkages are compared (Fig. 1 and Fig. S3, ESI†). Both the BLB- and LBL-type tetramers exhibit N and NTB phases. The LBL tetramer shows the higher TNI whereas BLB exhibits the higher stability of the NTB phase. This reinforces the view that the outer spacers are more important in controlling the overall molecular shape than the central spacer,25,26 such that the linear outer spacers promote nematic behaviour, and the bent outer spacers twist-bend nematic behaviour.
Birefringence measurements were performed for oligomeric compounds having the same, B-type linkages (Fig. 2 and Fig. S4, ESI†). The order parameter for flexible molecules may be related to the order of their mesogenic units. The optical birefringence, Δn, rises sharply at the transition from the isotropic to the nematic phase, and on cooling follows a critical dependence:  where Δnmax is the theoretical birefringence for the material with an ideal orientational order (order parameter, S = 1) and β is the critical exponent.27 Comparing Δnmax of the dimer CB6OCB, with that of the corresponding trimer CB6O.O6CB, and tetramer CB6O.6O.O6CB, it appears that the number of mesogenic units in the oligomer has only a minor effect on the birefringence, each showing Δnmax of around 0.3. The values of Δnmax of these oligomers are lower than that for the monomeric 5CB, for which Δnmax = 0.35,28 implying that in the oligomers the mesogenic units are at some angle with respect to the optical axis (director) of the nematic phase as a result of the overall bent-shape of these molecules. The birefringence decreases below the N–NTB transition because of the formation of the heliconical structure, and this decrease follows the increase of the conical angle.29 By analysing the departure from the critical behaviour, extrapolated to the temperature range of the NTB phase, the conical angle of the heliconical structure was determined to be in the range of 20–30 degrees at 50 K below the transition, this is in the range estimated using X-ray diffraction for other oligomers.30 Looking more closely at the birefringence behaviour, however, it is evident that Δn begins to depart from critical behaviour in the nematic phase a few degrees above the transition to the NTB phase. This arises from pretransitional fluctuations in the nematic phase, namely the formation of instantaneous helical structures.31 The decrease is most pronounced for the dimer, for the trimer the fluctuations are smaller and for the tetramer are very weak (Fig. 2 and Fig. S4 (ESI†), insets). This suggests that on increasing the number of mesogenic units that are linked together in the oligomer, the weaker these fluctuations become, and the discontinuity of the structure and N–NTB phase transition becomes stronger. This is also reflected in the associated entropy changes (Table S1, ESI†).
where Δnmax is the theoretical birefringence for the material with an ideal orientational order (order parameter, S = 1) and β is the critical exponent.27 Comparing Δnmax of the dimer CB6OCB, with that of the corresponding trimer CB6O.O6CB, and tetramer CB6O.6O.O6CB, it appears that the number of mesogenic units in the oligomer has only a minor effect on the birefringence, each showing Δnmax of around 0.3. The values of Δnmax of these oligomers are lower than that for the monomeric 5CB, for which Δnmax = 0.35,28 implying that in the oligomers the mesogenic units are at some angle with respect to the optical axis (director) of the nematic phase as a result of the overall bent-shape of these molecules. The birefringence decreases below the N–NTB transition because of the formation of the heliconical structure, and this decrease follows the increase of the conical angle.29 By analysing the departure from the critical behaviour, extrapolated to the temperature range of the NTB phase, the conical angle of the heliconical structure was determined to be in the range of 20–30 degrees at 50 K below the transition, this is in the range estimated using X-ray diffraction for other oligomers.30 Looking more closely at the birefringence behaviour, however, it is evident that Δn begins to depart from critical behaviour in the nematic phase a few degrees above the transition to the NTB phase. This arises from pretransitional fluctuations in the nematic phase, namely the formation of instantaneous helical structures.31 The decrease is most pronounced for the dimer, for the trimer the fluctuations are smaller and for the tetramer are very weak (Fig. 2 and Fig. S4 (ESI†), insets). This suggests that on increasing the number of mesogenic units that are linked together in the oligomer, the weaker these fluctuations become, and the discontinuity of the structure and N–NTB phase transition becomes stronger. This is also reflected in the associated entropy changes (Table S1, ESI†).
 | ||
Fig. 2 Birefringence vs. temperature for the tetramer CB6O.6O.O6CB (Δnmax = 0.298), measured for the wavelength 633 nm. The red line is the fit to the critical dependence  , the inset is a magnified section in the temperature range near the N–NTB transition. The blue curve represents the heliconical tilt angle θ in the NTB phase. , the inset is a magnified section in the temperature range near the N–NTB transition. The blue curve represents the heliconical tilt angle θ in the NTB phase. | ||
In non-resonant X-ray diffraction experiments, the diffuse signal located at 12–15 Å in the low angle region of the patterns seen for the N and NTB phases is related to the length of a single mesogenic unit, evidencing a locally intercalated structure. The resonant X-ray scattering studies, performed with the energy of the incident X-ray beam tuned to the carbon absorption edge, showed the diffraction signal in the NTB phase arising from the helical structure.32 On comparing the data for the dimer,10 trimer and tetramer it becomes evident that the helical pitch shortens on increasing the number of mesogenic units in the molecule, and is shortest for the tetramer (Fig. 3). It can also be seen that the temperature dependence of the pitch becomes weaker on increasing the number of mesogenic units linked in the oligomer. This temperature dependence of the helical pitch may be accounted for either in terms of increasing molecular tilt or by decreasing the number of mesogenic units forming the helix. We have seen that the weakest temperature dependence of the pitch in the NTB phase is observed for the tetramer and given that earlier it was shown that the tilt angle dependence for the dimer, trimer and tetramer is essentially the same, it appears that for the tetramer, the increase in tilt angle on lowering the temperature must be offset by additional mesogenic units being added to the helix. The weaker changes in the helical pitch near the NTB–N phase transition for the trimer and tetramer suggest that the N–NTB transition becomes more discontinuous on increasing the number of mesogenic units linked in the oligomer. This view is consistent with the earlier interpretation of the birefringence measurements.
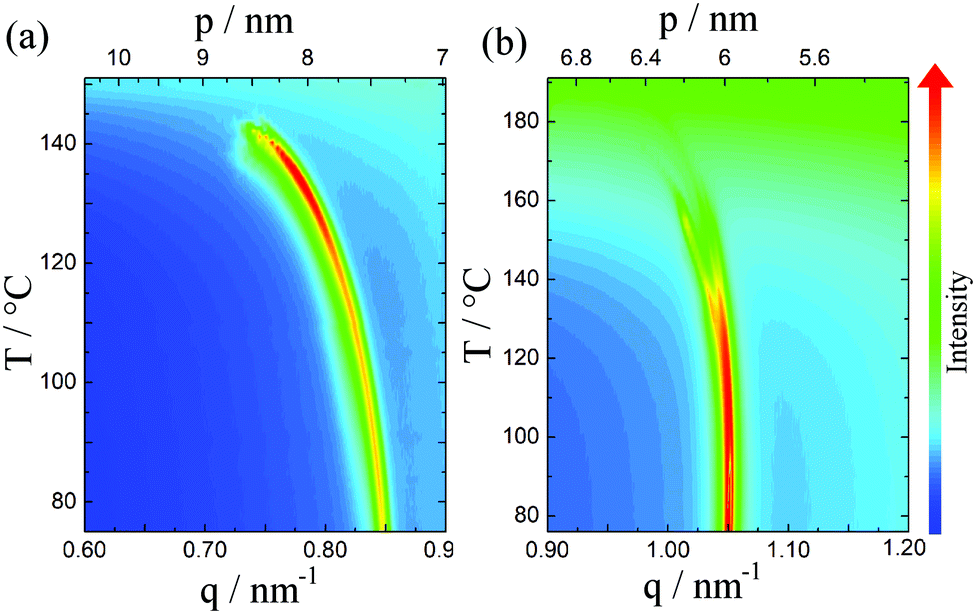 | ||
| Fig. 3 Resonant X-ray signal vs. temperature for the trimer CB6O.O6CB (a) and the tetramer CB6O.7.O6CB (b) measured at the carbon absorption edge. | ||
The helical pitch lengths measured using resonant X-ray diffraction correspond well to the distance between the stripes observed using AFM (atomic force microscopy) in the NTB phase (Fig. 4 and Fig. S5, ESI†). AFM studies have also shown that pitch of the helix strongly correlates to the mesogen arrangement in an oligomer, it is smallest for a BBB- and largest for a LBL-type structure (Fig. S5, ESI†). The LBL-type tetramer is additionally unstable in the NTB phase and crystallizes very easily and quickly.
The spontaneous formation of the heliconical structure in the nematic phase of bent molecules has been attributed to the anomalous behaviour of the bend elastic constant, K33; the formation of the NTB phase is driven by K33 tending towards negative values.33 For all the materials studied, the splay elastic constant, K11, increases monotonically on decreasing temperature, as a result of the increasing order parameter, and reaches a value of around 10 pN at approximately 20 K below the N-Iso phase transition (Fig. 5). In contrast, the bend elastic constant, K33, behaves non-monotonically, it initially increases below the N-Iso phase transition, but on further cooling decreases (Fig. 5), such behaviour has been previously reported for other dimeric mesogens exhibiting the NTB phase.34 This non-monotonic dependence of K33 on temperature is particularly strong for the trimer. This temperature dependence of K33 is a result of competing interactions: as the temperature is reduced, the orientational order increases and the value of K33 rises, at the same time, however, the tendency to form bent conformation increases which causes the decrease of K33. It is also evident that for the dimer and trimer, K33 starts to increase again in the close vicinity to the N–NTB phase transition, implying that the system becomes less susceptible to deformation when the instantaneous, local helical structure starts to form. No such increase is seen in K33 for the tetramer, and this is consistent with the other measurements showing that for this system the transition is strongly first order.
 | ||
| Fig. 4 AFM image obtained in the NTB phase for CB6O.8.O6CB compound (BLB-type). The helical pitch (distance marked by yellow arrows) is 12 nm. | ||
 | ||
| Fig. 5 Splay K11 (empty circles) and bend K33 elastic constants (solid circles) vs. temperature, measured in nematic phase for: the dimer CB6OCB (blue circles), the trimer CB6O.O6CB (black circles) and the tetramer CB6O.7.O6CB (red circles). | ||
This comparison of the behaviour of liquid crystalline oligomers has revealed that the stability of the heliconical NTB phase increases as the number of mesogenic units in the structure increases. For tetramers, the number and placement of the bend-imposing linkages in the structure is crucial. Introducing linear linkages destabilizes the NTB phase; notably, placement of L-type linkages in the outer fragments (LBL) has a much stronger effect than in the case of the central position (BLB). In addition, as the number of mesogenic units in the oligomer increases, the shorter the helical pitch becomes and it is less temperature dependent. The pitch length also depends on the sequence of the linear and bent fragments in the tetramer. The shortest pitch, in the range of 7 nm is seen for a BBB-type tetramer, and the longest for an LBL-type with a value of approximately 17 nm. The fluctuations in the nematic phase related to the formation of short range and instantaneous helices on approaching the N–NTB phase transition are less pronounced for the tetramer than for the trimer and in turn, those seen for the trimer are less pronounced than for the dimer. Thus, the N–NTB phase transition becomes more strongly first order in nature on increasing the number of mesogenic units in the molecular structure. This view is consistent with our calorimetric, birefringence and elastic constant measurements. It is very clear that we have much still to learn about the spontaneous chirality observed in these systems composed of achiral molecules, but this study of oligomeric structures shows how the pitch and its temperature dependence may be controlled by molecular structure. This has important implications for the potential use of these materials in areas such as nanotemplating and chiral separation techniques.
The work was financed by the National Science Centre (Poland) under the grant no. 2016/22/A/ST5/00319.
Conflicts of interest
There are no conflict of interest to declare.References
- T. Buhse, J. M. Cruz, M. E. Noble-Teran, D. Hochberg, J. M. Ribo, J. Crusats and J. C. Micheau, Chem. Rev., 2021, 121, 2147–2229 CrossRef CAS PubMed.
- C. Tschierske, Liq. Cryst., 2018, 45, 2221–2252 CrossRef CAS.
- M. Cestari, S. Diez-Berart, D. A. Dunmur, A. Ferrarini, M. R. De La Fuente, D. J. B. Jackson, D. O. Lopez, G. R. Luckhurst, M. A. Perez-Jubindo, R. M. Richardson, J. Salud, B. A. Timimi and H. Zimmermann, Phys. Rev. E: Stat., Nonlinear, Soft Matter Phys., 2011, 84, 31704 CrossRef CAS PubMed.
- V. Borshch, Y. K. Kim, J. Xiang, M. Gao, A. Jákli, V. P. Panov, J. K. Vij, C. T. Imrie, M. G. Tamba, G. H. Mehl and O. D. Lavrentovich, Nat. Commun., 2013, 4, 2635 CrossRef CAS PubMed.
- D. Chen, J. H. Porada, J. B. Hooper, A. Klittnick, Y. Shen, M. R. Tuchband, E. Korbloèa, D. Bedrov, D. M. Walba, M. A. Glaser, J. E. Maclennan and N. A. Clark, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 15931–15936 CrossRef CAS PubMed.
- C. Dressel, T. Reppe, M. Prehm, M. Brautzsch and C. Tschierske, Nat. Chem., 2014, 6, 971–977 CrossRef CAS PubMed.
- Y. Arakawa, K. Komatsu, T. Shiba and H. Tsuji, J. Mol. Liq., 2021, 326, 115319 CrossRef CAS.
- J. P. Abberley, R. Killah, R. Walker, J. M. D. Storey, C. T. Imrie, M. Salamończyk, C. Zhu, E. Gorecka and D. Pociecha, Nat. Commun., 2018, 9, 228 CrossRef PubMed.
- M. Salamończyk, N. Vaupotič, D. Pociecha, R. Walker, J. M. D. Storey, C. T. Imrie, C. Wang, C. Zhu and E. Gorecka, Nat. Commun., 2019, 10, 1922 CrossRef PubMed.
- M. R. Tuchband, D. A. Paterson, M. Salamończyk, V. A. Norman, A. N. Scarbrough, E. Forsyth, E. Garcia, C. Wang, J. M. D. Storey, D. M. Walba, S. Sprunt, A. Jákli, C. Zhu, C. T. Imrie and N. A. Clark, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 10698–10704 CrossRef CAS PubMed.
- R. J. Mandle and J. W. Goodby, Soft Matter, 2018, 14, 8846–8852 RSC.
- R. J. Mandle, M. P. Stevens & John and W. W. Goodby, Liq. Cryst., 2017, 44, 2046–2059 CrossRef CAS.
- F. P. Simpson, R. J. Mandle, J. N. Moore and J. W. Goodby, J. Mater. Chem. C, 2017, 5, 5102–5110 RSC.
- A. Al-Janabi, R. J. Mandle and J. W. Goodby, RSC Adv., 2017, 7, 47235–47242 RSC.
- R. J. Mandle, Soft Matter, 2016, 12, 7883–7901 RSC.
- R. J. Mandle and J. W. Goodby, Angew. Chem., 2018, 130, 7214–7218 CrossRef.
- R. J. Mandle and J. W. Goodby, ChemPhysChem, 2016, 17, 967–970 CrossRef CAS PubMed.
- Y. Arakawa, K. Komatsu, S. Inui and H. Tsuji, J. Mol. Struct., 2020, 1199, 126913 CrossRef CAS.
- R. Walker, D. Pociecha, A. Martinez-Felipe, J. M. D. Storey, E. Gorecka and C. T. Imrie, Cryst., 2020, 10, 175 CrossRef CAS.
- Y. Wang, G. Singh, D. M. Agra-Kooijman, M. Gao, H. K. Bisoyi, C. Xue, M. R. Fisch, S. Kumar and Q. Li, CrystEngComm, 2015, 17, 2778–2782 RSC.
- C. T. Imrie and G. R. Luckhurst, J. Mater. Chem., 1998, 8, 1339–1343 RSC.
- P. A. Henderson, A. G. Cook and C. T. Imrie, Liq. Cryst., 2004, 31, 1427–1434 CrossRef CAS.
- D. A. Paterson, J. P. Abberley, W. T. A. Harrison, J. Storey and C. T. Imrie, Liq. Cryst., 2017, 44, 127–146 CAS.
- D. A. Paterson, R. Walker, J. P. Abberley, J. Forestier, W. T. A. Harrison, J. M. D. Storey, D. Pociecha, E. Gorecka and C. T. Imrie, Liq. Cryst., 2017, 44, 2060–2078 CAS.
- T. Donaldson, P. A. Henderson, M. F. Achard and C. T. Imrie, J. Mater. Chem., 2011, 21, 10935–10941 RSC.
- P. A. Henderson and C. T. Imrie, Macromolecules, 2005, 38, 3307–3311 CrossRef CAS.
- I. Haller, Prog. Solid State Chem., 1975, 10, 103–118 CrossRef.
- J. Li and S. T. Wu, J. Appl. Phys., 2004, 96, 170 CrossRef CAS.
- C. Meyer, G. R. Luckhurst and I. Dozov, J. Mater. Chem. C, 2015, 3, 318–328 RSC.
- R. J. Mandle and J. W. Goodby, Phys. Chem. Chem. Phys., 2019, 21, 6839–6843 RSC.
- D. Pociecha, C. Crawford, D. A. Paterson, J. M. D. Storey, C. T. Imrie, N. Vaupotič and E. Gorecka, Phys. Rev. E, 2018, 98, 052706 CrossRef CAS.
- G. M. Su, I. A. Cordova, M. A. Brady, D. Prendergast and C. Wang, Polymer, 2016, 99, 782–796 CrossRef CAS.
- I. Dozov, Europhys. Lett., 2001, 56, 247–253 CrossRef CAS.
- G. Babakhanova, H. Wang, M. Rajabi, D. Li, Q. Li and O. D. Lavrentovich, Liq. Cryst. DOI:10.1080/02678292.2021.1973602.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1cc07012f |
| This journal is © The Royal Society of Chemistry 2022 |