
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Modulating the reaction pathway of phenyl diazonium ions using host–guest complexation with cucurbit[7]uril†
Suresh
Moorthy‡
a,
Alvaro
Castillo Bonillo‡
ab,
Hugues
Lambert
 ab,
Elina
Kalenius
c and
Tung-Chun
Lee
*ab
ab,
Elina
Kalenius
c and
Tung-Chun
Lee
*ab
aInstitute for Materials Discovery, University College London (UCL), London WC1H 0AJ, UK. E-mail: tungchun.lee@ucl.ac.uk
bDepartment of Chemistry, University College London (UCL), London WC1H 0AJ, UK
cDepartment of Chemistry, Nanoscience Center, University of Jyväskylä, Jyväskylä, Finland
First published on 17th February 2022
Abstract
Aryl diazonium ions are known to be an important intermediate in the divergent synthesis of azo compounds and substituted aromatics. The presence of more than one electrophilic center in a diazonium ion could lead to undesirable side reactions during a synthesis. Herein, we report that the electrophilic α-carbon on a phenyl diazonium [PhN2]+ ion can be selectively deactivated upon host–guest complexation with cucurbit[7]uril (CB7) in aqueous media, achieving a ∼60-fold increase in the half-life of [PhN2]+. Notably, however, the electrophilic nitrogen of the encapsulated diazonium ion remains active towards diazo coupling with strong nucleophiles, allowing the formation of azo compounds using a two-month-old aqueous solution of [CB7–PhN2]+. Our supramolecular approach can open new possibilities for the reactive chemistry of organic molecules in aqueous media.
Aryl diazonium compounds exhibit synthetic versatility as an intermediate for functional group interconversion of substituted aromatics, as well as the formation of azo dyes,1 such as azobenzene and its derivatives, which are the basic building blocks for a range of photoresponsive materials.2 A key feature that underpins the versatile chemistry of a diazonium ion is the presence of its two electrophilic reaction centers, namely the α-carbon and the N2 group. In particular, classically known as dediazoniation, the α-carbon typically reacts with a wide range of nucleophiles by displacing the N2 group via a SN1(Ar) (dissociative unimolecular nucleophilic aromatic substitution) or a SRN1(Ar) (dissociative radical nucleophilic aromatic substitution) mechanism.3 On the other hand, the N2 group is typically involved in diazo coupling reactions via the electrophilic aromatic substitution (SEAr) mechanism to form azo dyes.1
One potential downside arising from the multiple reactive centers within the diazonium functional group is the increased propensity of undesirable side reactions. In this context, a commonly encountered example is the reaction of diazonium ions with water, either as a solvent or from the atmospheric moisture, to form phenol. Indeed aryl diazonium chloride is only stable in water at up to 5 °C for a short period of time, and is therefore typically used in situ without isolation. Their stability can be increased by exchanging the counterions from halides to redox inactive and chaotropic anions, such as [BF4]−, which enable aryl diazonium salts to be isolated and even safely handled at elevated temperature for a short period of time.
Cucurbit[n]urils (CBn, n = 5–8) are a family of synthetic macrocyclic compounds that are chemically stable, water soluble and nontoxic.4,5 A CB molecule consists of a well-defined hydrophobic cavity with two symmetric carbonyl-group lined portals that are electron-rich. CBs can form host–guest complexes with small molecules driven by electrostatic interactions as well as classical and non-classical hydrophobic forces. They can recognize guests with a complementary shape, as illustrated by the isolation of o-xylene from its isomers.6 Meanwhile, CB7 is capable of forming a range of exceptionally strong complexes7 with binding constants rivalling and even exceeding that of the well-known biotin-avidin complex, one of the strongest noncovalent bonds found in nature.8
Interestingly, complexation with CBs can modulate the stability and, in some cases, the reactivity of the encapsulated guest molecule.9 For instance, the cavity of CBs can catalyze azide–alkyne Huisgen cycloaddition10 and Diels Alder reaction11via preorganization effects and enhancement of the local concentration, while van der Waals pulling by the cavity wall has been shown to facilitate the retro-Diels Alder reactions in the gas phase.12 The electron-rich portal of CBs, on the other hand, was shown to stabilize a cationic reaction intermediate of 2,2,6,6-tetramethylpiperidin-1-oxyl (TEMPO), which subsequently enables efficient biphasic oxidation of alcohols.13 Furthermore, the portal can also modulate the pKa of the encapsulated guest molecules by electrostatic effects, thereby allowing activation and stabilization of drugs in situ.14 Recently, the binding between CB7 and 4-nitrophenyldiazonium has been reported.15 Moreover it was shown that in the presence of CuCl, the complexation can promote decomposition of the guest into a mixture of 4-nitrobenzene (61%) and 4-nitrophenol (33%). Nevertheless, the mechanistic role of CB7 in aryl diazonium chemistry remains unknown.
Herein, we report the modulation of the reaction pathway of phenyl diazonium [PhN2]+ ions via site-specific deactivation using host–guest complexation with CB7 (Fig. 1). In particular, we show that encapsulation of a [PhN2]+ ion inside CB7 can effectively deactivate the α-carbon site towards SN1(Ar) reaction with water molecules, resulting in a ∼60-fold increase in stability (half-life t1/2 = 1.30 × 106 s, i.e. 15 days) compared to free [PhN2]+ ions in aqueous media at 25 °C, which represents the first example of lifetime enhancement of aqueous diazonium ions via supramolecular means. Computational modelling of transition states indicates that deactivation of the α-carbon site can be attributed to the electrostatic interactions between the cationic diazonium group and the CB7 portal. Notably, however, the N2 group of an encapsulated [PhN2]+ ion remains active towards diazo coupling with strong nucleophiles via the SEAr pathway, as the N2 group protrudes slightly further away from the CB7 portal. The combined reactivity modulation effects allow the formation of azo compounds using a two-month-old aqueous solution of [CB7–PhN2]+.
 | ||
| Fig. 1 Schemes of (a) typical [PhN2]+ reactive pathways via SN1(Ar) and SEAr mechanisms, and (b) modulated pathways upon encapsulation of the [PhN2]+ ion by CB7, where the SN1 route is effectively hindered. | ||
We began our investigation into the potential role of CB7 in modulating aryl diazonium chemistry by studying the well-established SN1(Ar) reaction of [PhN2]+ ions with water molecules.3 In particular, the reaction readily occurs in aqueous media at room temperature, as indicated by the decreasing intensity of the UV-vis absorption peak at ∼260 nm and ∼300 nm (Fig. S1 and S2, ESI†). Despite the convenience of UV-vis spectroscopy, we observed that UV irradiation can increase the rate of decomposition (Fig. S3–S5, ESI†). We therefore turned to non-optical techniques, particularly 1H NMR, for tracking the reaction kinetics.
Evolution of the 1H NMR signals of 10 mM [PhN2]+[BF4]− in D2O contained in a brown NMR tube was measured by in situ1H NMR over the course of 30 hours, as shown in Fig. S6 (ESI†). At time = 0, three NMR peaks (δ = 8.58, 8.31 and 7.98 ppm) can be observed which correspond to the three proton environments in a [PhN2]+ ion. As time progresses, the NMR signals from [PhN2]+ ions weaken while a new set of three NMR peaks (δ = 7.32, 6.99 and 6.92 ppm), which corresponds to phenol, gradually grows. Given the known initial concentration of [PhN2]+ and the linear correlation between concentration and total area under the NMR peaks, we can plot the absolute concentration of [PhN2]+ against time (Fig. S7, ESI†). The linearity in the plot of ln(concentration of [PhN2]+) against time confirms that the reaction obeys the first order rate law as expected (Fig. S8, ESI†), and reveals the reaction rate constant k to be 3.19 × 10−5 s−1 at 25 °C with a corresponding half-life t1/2 of [PhN2]+ = 2.17 × 104 s, i.e. 0.25 days. Further measurements also confirm that the reaction rate is practically pH-independent in the pH range of our work (Fig. S9, ESI†).
Host–guest complexes of CB7 and [PhN2]+ readily form via aqueous self-assembly, upon mixing of the corresponding sample solutions. The encapsulation of a [PhN2]+ ion by the CB7 cavity is evident by the up-field shift of its NMR peaks as shown in Fig. 2a. In particular, the significant upfield shift of Hb (Δδ = 0.94 ppm) indicates that it resides deep inside the CB7 cavity, while the less notable shift of Ha and Hc suggests that they sit slightly closer to the deshielding region of the carbonyl portal,16 which is consistent with the energy-minimized molecular model of the 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 host–guest complex (Fig. 2b). Further support for the 1:1 binding stoichiometry was found in the mass spectra of the host–guest complex sample measured by electrospray ionization mass spectrometry (ESI-MS) as shown in Fig. S10 and S11 (ESI†). In particular, the mass spectra showed 1:1 complex [CB7–PhN2 + H]2+ at m/z 634.2 (Table S1, ESI†). Collision cross section (CCS) values were defined using ion mobility mass spectrometry. DTCCSN2 of [CB7–PhN2 + H]2+ (337.8 Å2; DTCCSN2 for [CB7 + 2H]2+ is 334.7 Å2) indicates encapsulation of [PhN2]+ deep inside the CB7 cavity. We note that it has not been possible to precisely extract the binding constant using UV-vis or NMR titrations due to chemical instability of [PhN2]+ ions in aqueous media. See Section S6 for a further discussion about the binding constant and kinetics (ESI†).
:1 host–guest complex (Fig. 2b). Further support for the 1:1 binding stoichiometry was found in the mass spectra of the host–guest complex sample measured by electrospray ionization mass spectrometry (ESI-MS) as shown in Fig. S10 and S11 (ESI†). In particular, the mass spectra showed 1:1 complex [CB7–PhN2 + H]2+ at m/z 634.2 (Table S1, ESI†). Collision cross section (CCS) values were defined using ion mobility mass spectrometry. DTCCSN2 of [CB7–PhN2 + H]2+ (337.8 Å2; DTCCSN2 for [CB7 + 2H]2+ is 334.7 Å2) indicates encapsulation of [PhN2]+ deep inside the CB7 cavity. We note that it has not been possible to precisely extract the binding constant using UV-vis or NMR titrations due to chemical instability of [PhN2]+ ions in aqueous media. See Section S6 for a further discussion about the binding constant and kinetics (ESI†).
 | ||
| Fig. 2 Formation of [CB7–PhN2]+ inclusion host–guest complexes. (a) Stack plot of 1H NMR spectra of CB7 (bottom), [PhN2]+[BF4]− (middle) and a 1:1.5 mixture of [PhN2]+[BF4]− and CB7 (top) in D2O at 25 °C monitored using a 400 MHz NMR instrument, which confirms the characteristic upfield shift of the NMR peaks of [PhN2]+ upon host–guest complexation. (b) Molecular model of [CB7–PhN2]+ optimized at the CPCM/wB97XD/6-31G* level of theory, supporting the NMR peak assignment. | ||
Notably, the encapsulation of [PhN2]+ by CB7 effectively slows down its reaction with water, despite the fact that the α-carbon site remains accessible by water molecules after complexation. In situ NMR measurement in D2O over 37 days confirms that the reaction obeys the first-order rate law (Fig. 3 and Fig. S6–S8, ESI†), and reveals the reaction rate constant k to be 5.35 × 10−7 s−1 at 25 °C with a corresponding half-life t1/2 of [CB7–PhN2]+ = 1.30 × 106 s, i.e. 15 days, which represents a 60-fold increase and a ΔΔG‡ of +2.4 kcal mol−1 with respect to free [PhN2]+. Despite precedent reports on binding between diazonium ions and supramolecular hosts (e.g. CB7,15 cyclodextrin,17 crown ether18–20 and metal–organic cages21), our finding features the first example of enhancing the lifetime of reactive aqueous diazonium ions via a supramolecular approach. Phenol was formed as the product of the reaction and existed as host–guest complexes with CB7, as evident by the emerging 1H NMR signals in the range of 6.90–6.10 ppm (Fig. 3a). The identity of the reaction product was verified by further NMR measurements (Fig. S13, ESI†). Meanwhile it is noted that a trace amount of benzene (yield <6%) was formed as the side product (Fig. S14, ESI†).
 | ||
| Fig. 3 SN1 reaction kinetics of [PhN2]+ and [CB7–PhN2]+. (a) 1H NMR spectra of a mixture of CB7 (6.2 mM) and [PhN2]+ (4.0 mM) and its evolution over time. (b) First-order kinetics plot of [PhN2]+ and [CB7–PhN2]+ samples. Transition state model of [CB7–PhN2–H2O]+ at the wB97XD/6-31G* level of theory; (c) top and (d) side views. Part of the CB7 framework is hidden for an unobstructed view of the encapsulated guest. Electrostatic partial charges are shown for selected atoms involved in the SN1 reaction. | ||
To gain a fundamental understanding of the α-carbon deactivation by CB7, we performed computational modelling based on dispersion-corrected methods (see ESI,† Section S1d and S8 for details), which were chosen to estimate the van der Waals interactions that contribute to the stability of the host–guest complexes. The binding energy of the [CB7–PhN2]+ complex (Fig. 2b) was computed to be −4.27 kcal mol−1, using the tight-binding method GFN2-xTB with the alpb solvation model and thermal contributions, which is comparable to but weaker than that between CB7 and protonated aniline.
Theoretical kinetic parameters of the reaction, in particular the Gibbs free energy of activation (ΔG‡ = ΔH‡ − TΔS‡, where ΔH‡ and ΔS‡ are enthalpy and entropy of activation, respectively, and T is the absolute temperature), can be extracted from the transition state models of the [CB7–PhN2–H2O]+ complex (Fig. 3c and d) and the free [PhN2–H2O]+ within a solvent cage of explicit water molecules (Fig. S15, ESI†). The DFT method CPCM/wB97XD/6-31G* was used due to its proven track record in calculating energy barriers. Although absolute values for energy barriers can suffer errors of up to several kcal mol−1, we expect a degree of error cancelation to yield relatively accurate relative energy barrier differences.
In particular, ΔH‡ increases from +28.02 to +31.46 kcal mol−1 (ΔΔH‡ = +3.44 kcal mol−1) upon complexation with CB7, which can be attributed to the following reasons. As indicated by the transition state models and intrinsic reaction coordinate (IRC) analysis (Fig. S16, ESI†), upon formation of the transition state, the positive electrostatic charge density on the N2-group gradually shifts towards the α-carbon site on the phenyl ring, resulting in species that resemble a phenyl carbocation and a dinitrogen molecule. In the case of a CB7 complex, the shift in charge density towards the cavity weakens its electrostatic interactions with the CB7 portal. In addition, the cavity of CB7 exhibits an exceptionally low dielectric constant because all electron lone pairs on CB7 are pointing outwards.9 As a result, the positively charged phenyl ring is not effectively stabilized by ion-induced dipole interactions in the CB7 cavity compared to within a solvent cage of water (see Fig. S15 for computed partial charges, ESI†). It is noted that the presence of a [BF4]− counterion around the [CB7–PhN2]+ complex only shows insignificant effects on the partial charges of the diazonium moiety (Fig. S17, ESI†), and therefore is not expected to contribute to modulating the reactivity of the CB7-encapsulated [PhN2]+.
On the other hand, we estimated the entropic term TΔS‡ by considering the configurational entropy extracted from the frequencies calculation of the optimized DFT model at the same level of theory. The TΔS‡ at 300 K increases from +0.88 to +0.92 kcal mol−1 (TΔΔS‡ = −0.04 kcal mol−1) upon complexation. The small theoretical TΔS‡ and TΔΔS‡ imply that configurational entropy does not play any major role in this particular SN1(Ar) kinetics.
Overall, the formation of host–guest complexes increases the theoretical ΔG‡ from +27.14 to +30.54 kcal mol−1 with ΔΔG‡ = +3.4 kcal mol−1, which matches well with the experimental ΔΔG‡ of +2.4 kcal mol−1. The higher free energy barrier implies that phenyl diazonium ions become more kinetically stable inside the CB7 cavity against the SN1(Ar) reaction with water. Since the rate determining step of the SN1(Ar) mechanism is the formation of a carbocation, the observed deactivation of the α-carbon on [PhN2]+ is expected to be also applicable to other weak nucleophiles, e.g. chloride. It is noted that, in the presence of CB7, the SN1(Ar) reaction with water could proceed either within the [CB7–PhN2]+ complex via a higher kinetic barrier, or in the free [PhN2]+ form during the fast host–guest exchange events, which is thermodynamically disfavored but kinetically more viable. Sophisticated computational models, e.g. infrequent metadynamics,22 are needed for deeper insights.
Strikingly, despite the effective deactivation of the α-carbon, we discovered that the N2-group of a [CB7–PhN2]+ complex remains active towards diazo coupling reactions, as illustrated by the diazo coupling reaction with 2-naphthoxide ions using aged samples of [PhN2]+ and [CB7–PhN2]+ in water (Fig. 4 and see ESI,† Section S9 for details). In particular, for freshly prepared [PhN2]+ and [CB7-PhN2]+ samples, i.e. Δt = 0, both samples rapidly produced the azo dye (Sudan I) upon addition of excess 2-naphthoxide ions. Extraction and quantification of Sudan I using UV-vis spectroscopy indeed confirmed that the diazo coupling reaction of the [CB7–PhN2]+ complexes proceeded without any notable hindrance (yield = 92%) with respect to that of the [PhN2]+ ions (yield = 94%). For samples aged for 1 day, diazo coupling was observed for [CB7–PhN2]+ complexes with a yield as high as 77%, whereas the reaction yield in the absence of CB7 rapidly dropped to 6%. Interestingly, Sudan I formation can be observed in a [CB7–PhN2]+ sample that has been aged in water for 70 days (yield 3%). The retainment of the N2-group reactivity in [CB7–PhN2]+ complexes can be attributed to the N2 group being protruded slightly further away from the CB7 portal (Fig. 2b) and therefore unaffected by the host–guest complexation. Furthermore, the observed regioselectivity (SN1(Ar) vs. SEAr) is shown to be generic across other nucleophilic substrates, including cresols and aniline (Fig. S19, ESI†).
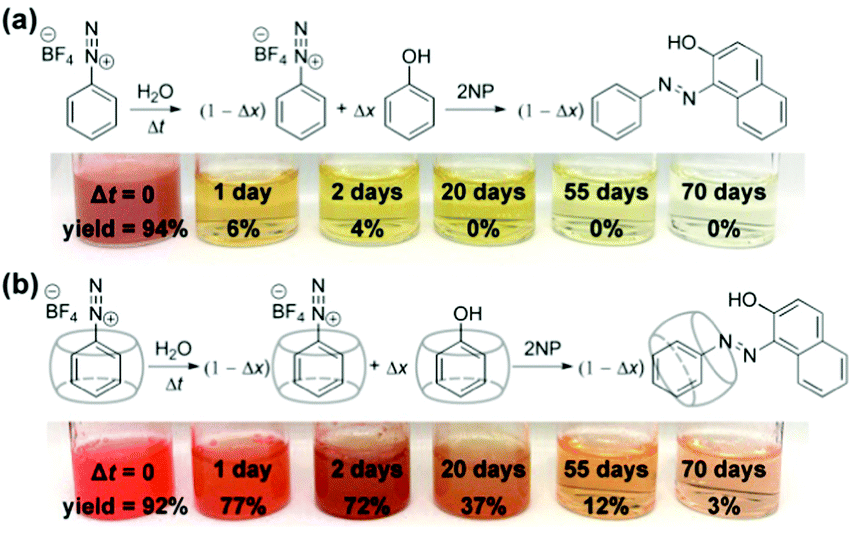 | ||
| Fig. 4 Bimolecular SEAr reactivity of (a) [PhN2]+ and (b) [CB7–PhN2]+ in water over time. Photographs showing resultant reaction mixtures prepared by adding 2-naphthoxide (2NP) into free [PhN2]+ (1.3 mM) and the mixture of [PhN2]+ (1.3 mM) and CB7 (1.9 mM) solutions aged for 0, 1, 2, 20 and 55 and 70 days. Corresponding % yields are also shown. | ||
We report the host–guest complexation between CB7 and phenyl diazonium ions in aqueous media, which represents a relatively rare example of CB binding to a highly reactive guest molecule. Moreover, our results show that the complexation can selectively modulate the reaction pathway of the encapsulated guest via deactivation of the α-carbon site while keeping the reactivity of the N2-group intact. Our findings represent the first example of lifetime enhancement of aqueous diazonium ions via supramolecular means, featuring a ∼60-fold increase in stability (half-life t1/2 = 1.30 × 106 s, i.e. 15 days) compared to free [PhN2]+ ions in aqueous media at 25 °C. Computational modelling indicates that CB7 can electrostatically destabilize the transition state of the SN1(Ar) pathway. Interestingly, the N2-group of a [CB7–PhN2]+ complex remains active towards diazo coupling reaction as illustrated by using [CB7–PhN2]+ samples that were aged in water for as long as 70 days. Site-specific reactivity modulation, especially to distinguish different reactive centers within the same functional group, using artificial supramolecular hosts is rare and challenging to realize.23 Our supramolecular approach can potentially be extended to modulate reaction pathways of other small organic molecules, and open new possibilities for reactive chemistry of organic molecules in aqueous media.
This work is funded by the Leverhulme Trust (RPG-2016-393). A. C. B. is grateful to the A*STAR-UCL Studentship funded via the EPSRC M3S CDT (EP/L015862/1). We acknowledge the use of the UCL Myriad high performance computing facility, and the UK Materials and Molecular Modelling Hub, which are partially funded by EPSRC (EP/P020194/1).
Conflicts of interest
There are no conflicts to declare.Notes and references
- E. Merino, Chem. Soc. Rev., 2011, 40, 3835–3853 RSC.
- K. Rafal, Pure Appl. Chem., 2010, 82, 2247–2279 Search PubMed.
- Z. Wu and R. Glaser, J. Am. Chem. Soc., 2004, 126, 10632–10639 CrossRef CAS PubMed.
- S. J. Barrow, S. Kasera, M. J. Rowland, J. del Barrio and O. A. Scherman, Chem. Rev., 2015, 115, 12320–12406 CrossRef CAS PubMed.
- J. Kim, I.-S. Jung, S.-Y. Kim, E. Lee, J.-K. Kang, S. Sakamoto, K. Yamaguchi and K. Kim, J. Am. Chem. Soc., 2000, 122, 540–541 CrossRef CAS.
- G. Zhang, A.-H. Emwas, U. F. S. Hameed, S. T. Arold, P. Yang, A. Chen, J.-F. Xiang and N. M. Khashab, Chemistry, 2020, 6, 1082–1096 CrossRef CAS.
- H. Lambert, N. Mohan and T.-C. Lee, Phys. Chem. Chem. Phys., 2019, 21, 14521–14529 RSC.
- M. V. Rekharsky, T. Mori, C. Yang, Y. H. Ko, N. Selvapalam, H. Kim, D. Sobransingh, A. E. Kaifer, S. Liu, L. Isaacs, W. Chen, S. Moghaddam, M. K. Gilson, K. Kim and Y. Inoue, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 20737 CrossRef CAS PubMed.
- K. I. Assaf and W. M. Nau, Chem. Soc. Rev., 2015, 44, 394–418 RSC.
- W. L. Mock, T. A. Irra, J. P. Wepsiec and T. L. Manimaran, J. Org. Chem., 1983, 48, 3619–3620 CrossRef CAS.
- A. Palma, M. Artelsmair, G. Wu, X. Lu, S. J. Barrow, N. Uddin, E. Rosta, E. Masson and O. A. Scherman, Angew. Chem., Int. Ed., 2017, 56, 15688–15692 CrossRef CAS PubMed.
- T.-C. Lee, E. Kalenius, A. I. Lazar, K. I. Assaf, N. Kuhnert, C. H. Grün, J. Jänis, O. A. Scherman and W. M. Nau, Nat. Chem., 2013, 5, 376–382 CrossRef CAS PubMed.
- Y. Jiao, B. Tang, Y. Zhang, J.-F. Xu, Z. Wang and X. Zhang, Angew. Chem., Int. Ed., 2018, 57, 6077–6081 CrossRef CAS PubMed.
- N. i. Saleh, A. L. Koner and W. M. Nau, Angew. Chem., Int. Ed., 2008, 47, 5398–5401 CrossRef CAS PubMed.
- H. Xu and Q. Wang, Chin. Chem. Lett., 2019, 30, 337–339 CrossRef CAS.
- W. L. Mock and N. Y. Shih, J. Org. Chem., 1986, 51, 4440–4446 CrossRef CAS.
- A. Fernández-Alonso and C. Bravo-Díaz, J. Colloid Interface Sci., 2012, 368, 301–309 CrossRef PubMed.
- R. A. Bartsch and P. N. Juri, J. Org. Chem., 1980, 45, 1011–1014 CrossRef CAS.
- E. P. Kyba, R. C. Helgeson, K. Madan, G. W. Gokel, T. L. Tarnowski, S. S. Moore and D. J. Cram, J. Am. Chem. Soc., 1977, 99, 2564–2571 CrossRef CAS.
- G. W. Gokel and D. J. Cram, J. Chem. Soc., Chem. Commun., 1973, 481–482 RSC.
- J. L. Brumaghim, M. Michels, D. Pagliero and K. N. Raymond, Eur. J. Org. Chem., 2004, 5115–5118 CrossRef CAS.
- H. Lambert, Y.-W. Zhang and T.-C. Lee, J. Phys. Chem. C, 2020, 124, 11469–11479 CrossRef CAS PubMed.
- M. Yoshizawa, J. K. Klosterman and M. Fujita, Angew. Chem., Int. Ed., 2009, 48, 3418–3438 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1cc06982a |
| ‡ Authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2022 |