
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHeterogeneity of oxygen reactivity: key for selectivity of partial methanol oxidation on gold surfaces†
Christoph D.
Feldt
 ,
Paul A.
Albrecht
,
Salma
Eltayeb
,
Wiebke
Riedel
* and
Thomas
Risse
*
,
Paul A.
Albrecht
,
Salma
Eltayeb
,
Wiebke
Riedel
* and
Thomas
Risse
*
Institut für Chemie und Biochemie, Freie Universität Berlin, Arnimallee 22, 14195 Berlin, Germany. E-mail: wiebke.riedel@fu-berlin.de; risse@chemie.fu-berlin.de
First published on 9th March 2022
Abstract
Recent evidence for low-temperature oxidation of methyl formate on Au(332) may affect the selectivity of gold catalysts during partial oxidation of methanol. Under isothermal conditions, overoxidation of methyl formate is significantly slower than methanol oxidation which can be attributed to special oxygen species required for overoxidation.
Methyl formate is an important intermediate for a variety of bulk chemicals, such as formic acid, formamide or dimethyl formamide, and more recently was also tested as a possible alternative fuel (additive).1–4 Apart from the currently applied industrial synthesis (reaction of CO with methanol over sodium or potassium methoxide requiring dry and CO2-free feeds),2,4,5 dehydrogenation and the thermodynamically more favourable partial oxidation of methanol are pursued as alternative routes.1,2 On nanoporous gold (np-Au), aerobic partial oxidation of methanol to methyl formate shows a temporally stable, high selectivity at high conversion for temperatures below 100 °C.6 Np-Au is a fully metallic catalyst which is prepared by dealloying a gold alloy containing a less noble metal, mostly Ag, resulting in a porosity on the nanometer scale characterized by ligaments exhibiting a large fraction of low coordinated sites as well as low index facets.7–9 Residual Ag in np-Au was found to be key for aerobic oxidation catalysis. The activation of molecular oxygen is typically the rate limiting step8,10 rendering the transient concentration of activated oxygen on np-Au low under typical reaction conditions.11,12 Increasing the reactive oxygen concentration either by increasing the Ag content or the oxygen pressure resulted in a reduced selectivity towards methyl formate formation.6,11,13 In agreement, surface science studies employing temperature programmed reaction (TPR) measurements on Au(111) showed preferred formation of methyl formate at low coverage of activated oxygen, while high coverages of activated oxygen favour total oxidation of methanol.14 Please note, that molecular oxygen does not dissociate on Au surfaces under ultrahigh vacuum (UHV) conditions which requires supply of activated oxygen (O-atoms, ozone etc.) in UHV experiments.15,16 A recent TPR study on Au(332), a surface with 6 atom-wide (111)-terraces separated by monoatomic steps as well as a smaller number of kink sites depending on the miscut of the crystal,17 showed oxidation of methyl formate already at 135 K. This was observed for oxygen coverages previously reported to result in high selectivity to methyl formate formation from methanol on Au(111).18 In addition, the overoxidation is found well-below the temperature of methyl formate formation reported for the TPR experiments on methanol oxidation mentioned above which shows that the activation energy of the former reaction is low enough to allow for an overoxidation of methyl formate on stepped gold surfaces.18 While methyl formate oxidation may not occur for methanol oxidation in TPR experiments, the presence of low-temperature overoxidation channels might be important for the selectivity of isothermal catalysis on np-Au, where molecules will exhibit multiple collisions with the catalyst surface along the catalyst bed. While temperature programmed experiments allow to identify such reaction channels, conclusions on their impact on the isothermal kinetics of the reaction network, hence, the selectivity of a catalyst can typically not be drawn. This requires isothermal experiments. We will present an isothermal reactivity study under well-defined conditions on a Au(332) single crystal surface after discussing additional TPR experiments on this system.
TPR measurements of methyl formate adsorbed at low temperature on oxygen pre-covered Au(332) and subsequently heated (see Fig. 1a and ESI† for experimental details) reveal the formation of CO2 at 135 K, 185 K and 320 K, based on the lack of CO2 related signals in reference measurements. Using isotopic labelling, CO2 formation at these temperatures was attributed to three distinct reaction pathways consistent with an attack of oxygen on the carbonyl carbon, the methyl carbon and through a cyclic transition state, respectively.18 Accordingly, a different and rather specific geometry of the transition state is expected to be required for all three pathways.18 Moreover, the CO2 desorption at 320 K resulted from dehydrogenation of formate species which are formed at low temperatures and are located on special sites, presumably kinks.18 It should be noted that the observed desorption of unreacted methyl formate is consistent with a sufficiently high coverage to prevent restricted CO2 formation by low temperature availability of methyl formate (see also Fig. S1, ESI†). For high pre-coverages of activated oxygen, however, the CO2 desorption features at 135 K and 320 K vanish (see Fig. 1b). CO2 desorption at 185 K coincides with methyl formate desorption hampering a quantitative analysis of the CO2 amount for this pathway. Compared to the signals at 135 K and 320 K for low oxygen coverage, however, only a small amount of CO2 desorbs at 185 K. For CO2 desorption at 135 K and 320 K, Fig. 1c evidences a steep drop in the amount of CO2 for increasing oxygen pre-coverages indicative of a blocking of corresponding sites by excess oxygen. Thus, methyl formate oxidation requires a small surface coverage of activated oxygen. Please note, the oxygen coverage is also low for typical reaction conditions on np-Au catalysts11,12 which renders such reaction channels potentially important. It should be noted that the decrease in CO2 formation on Au(332) occurs when the oxygen pre-coverage exceeds the number of step sites on Au(332) (approx. 17%). As theoretical calculations indicate a preferential decoration of low-coordinated sites (steps, kinks, etc.) by oxygen atoms,19 it is concluded that oxygen species active in methyl formate oxidation are predominantly found at low coordinated sites. It is estimated for the lowest oxygen pre-coverage (0.13 ML) that approx. one CO2 molecule is formed per reacted methyl formate molecule (see ESI† for details), in agreement with previous isotopic labelling results.18 For the oxygen pre-coverage of 0.13 ML associated with the highest observed CO2 formation, less than 10% of the oxygen is used to form CO2 in all three reaction pathways combined (see ESI† for details). This is significantly lower than the number of step sites on the Au(332) surface (approx. 17%). The estimation is consistent with desorption of unreacted oxygen (and methyl formate) observed for all TPR measurements. These results evidence that all three reaction pathways resulting in methyl formate oxidation require special oxygen species on low coordinated sites which exhibit a low apparent activation barrier as indicated by the CO2 (or formate) formation already at temperatures below 200 K (see Fig. 1 and ref. 18). While these experiments clearly identify overoxidation channels of methyl formate at low temperature and low oxygen coverage, the importance of TPR experiments for the selectivity under isothermal conditions cannot be judged.
 | ||
| Fig. 1 C16O2 desorption of TPR experiments with 0.11 L methyl formate (black) on Au(332), pre-covered with (a) 0.13 ML and (b) 1.3 ML 16O. For comparison, also results of reference measurements of the O pre-covered Au(332) surface are shown (grey). (c) Integrated intensity of CO2 evolution at 135 K (blue) and 320 K (red) in TPR experiments of 0.11 L methyl formate on oxygen pre-covered Au(332) with O pre-coverages ranging between 0.13 ML and 1.3 ML. Error bars are estimated from the area variation obtained for different integration approaches (see ESI† for details). | ||
To elucidate this question, isothermal pulsed molecular beam (MB) experiments were performed on Au(332) which ensure single collision conditions, thus, preventing multiple collisions of educts and reaction products with the surface. In turn, the fluxes of methyl formate and methanol colliding with the surface can be controlled which allows to study the competition of the respective oxidation reactions under isothermal conditions. In the experiment shown in Fig. 2a, the surface was continuously exposed to methyl formate, while oxygen atoms (18O) were pulsed for 30 s at a low flux (approx. 0.015 ML per pulse; flux 6.9 × 1011 cm−2 s−1) onto the surface (see also ESI† for experimental details). The applied surface temperature of 310 K was chosen to be similar to those reported in np-Au studies6 and sufficiently high to prevent surface deactivation by formate accumulation, i.e. above the onset of the high-temperature CO2 desorption exhibiting a maximum at 320 K (see Fig. 1a). Formation of water (H218O) during the oxygen pulse demonstrates methyl formate oxidation to proceed on Au(332) also under isothermal conditions. It should be noted that detection of other (potential) products including CO2 is impeded by strong fragmentation signals of methyl formate or non-negligible reactions in the chamber exposed to atomic oxygen, as detected by reference measurements applying a non-reactive quartz flag. Upon atomic oxygen exposure (dashed line at 0 s), the H218O rate increases within approx. 10 s before levelling to a constant rate (approx. 30 counts per second (cps)). After the end of the oxygen pulse (dashed line at 30 s), the rate remains constant for at least 20 s before decaying back to the level prior to the oxygen pulse. This behaviour is consistent with accumulation of some of the oxygen during the pulse which is quantitatively consumed in the delay time. This is further supported by the absence of 18O2 desorption in a temperature programmed desorption (TPD) measurement after the pulsed MB experiment (see Fig. S2, ESI†). The oxygen accumulation during the oxygen pulse indicates a rather slow reaction of oxygen with methyl formate on Au(332) considering the large excess of methyl formate flux (factor of approx. 1700) as compared to that of oxygen atoms.
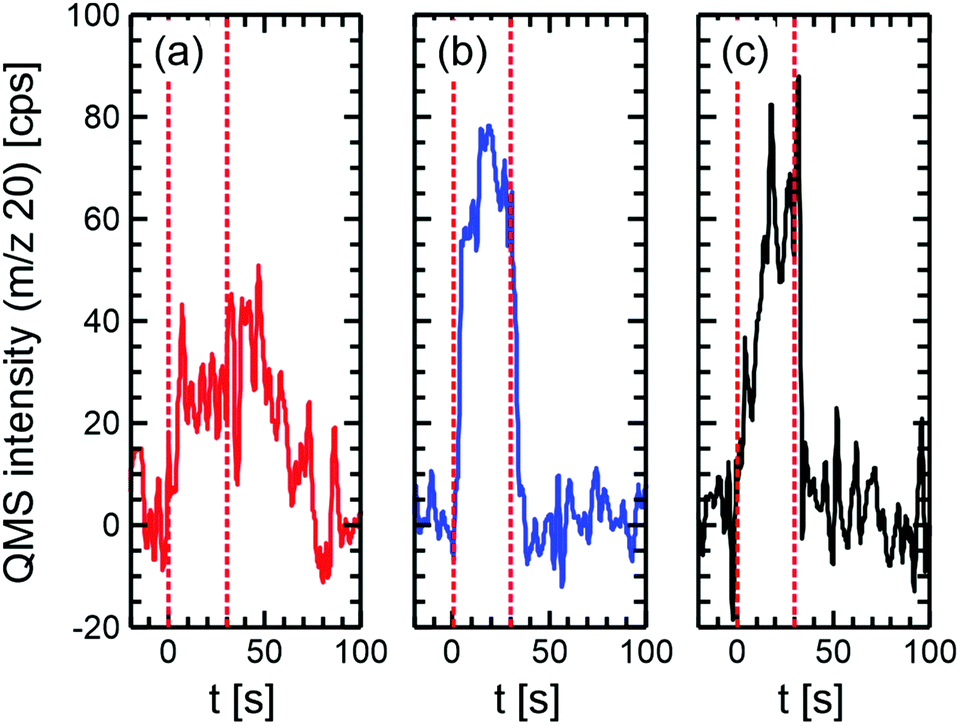 | ||
| Fig. 2 Isothermal pulsed molecular beam experiments on Au(332) at 310 K pulsing atomic oxygen (6.9 × 1011 cm−2 s−1, 30 s on, 100 s off), while continuously supplying (a) methyl formate (1.2 × 1015 cm−2 s−1), (b) methanol-13C (4.2 × 1013 cm−2 s−1) and (c) both methanol-13C (4.2 × 1013 cm−2 s−1) and methyl formate (1.2 × 1015 cm−2 s−1) at a large excess in the gas phase. The graphs show the difference of the H218O (m/z = 20) production rate (averaged over 31 pulses) between exposed Au(332) and a reference measurement with a non-reactive quartz flag. The length of one single oxygen pulse is indicated by the red dashed lines. | ||
In comparison to that, oxidation of methanol-13C (60-fold excess of flux compared to oxygen) shows a steep increase in the H218O formation upon oxygen exposure (same condition as above; approx. 0.015 ML per pulse) levelling off at about twice the rate and returning back to baseline shortly after the end of the oxygen pulse indicating almost quantitative consumption of oxygen during the pulse (see Fig. 2b; for 18O2-TPD after the reaction see Fig. S2, ESI†). It should be noted that no methyl formate formation is detected under the applied conditions for temperatures above 270 K,20,21 as formaldehyde desorption competes effectively at high temperatures with the coupling reaction under single collision conditions, similar to short contact times in np-Au catalysts.11 Detection of small amounts of formaldehyde is, however, hampered by large (fragmentation) signals due to methanol. These results suggest a significantly faster reaction of methanol with oxygen on Au(332) as compared to the methyl formate oxidation, despite the 30-fold reduced flux of methanol as compared to that of methyl formate.
For a direct comparison, a pulsed MB experiment with a co-feed of methanol and methyl formate using the fluxes of the individual experiments presented above (30-fold excess of methyl formate) was conducted (Fig. 2c). Please note that such a flux ratio of 30![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 corresponds to a gas mixture in np-Au at conversion well above 90%. In this measurement, the quasi-steady-state H218O formation rate is only slightly lower than that observed for methanol oxidation, in agreement with a significantly faster methanol than methyl formate oxidation on Au(332) despite the higher flux of methyl formate. However, the slight reduction in the quasi steady state rate as well as a slower increase and decrease after start and end of the oxygen exposure, respectively, show a kinetically relevant contribution of methyl formate to the observed water formation. The reaction probability of oxygen towards water can be estimated to be lower by a factor of approx. 60 for methyl formate than for methanol considering the quasi-steady-state rate during the pulse is halved (Fig. 2a and b) and the flux is increased by a factor of approx. 30 for methyl formate (combined with quantitative oxygen consumption by the end of the delay time). This strong reactivity difference suggests gold surfaces allowing for high methanol conversions, before overoxidation of the partial oxidation product methyl formate significantly contributes to a selectivity loss, as desired for an ideal partial oxidation catalyst.
:1 corresponds to a gas mixture in np-Au at conversion well above 90%. In this measurement, the quasi-steady-state H218O formation rate is only slightly lower than that observed for methanol oxidation, in agreement with a significantly faster methanol than methyl formate oxidation on Au(332) despite the higher flux of methyl formate. However, the slight reduction in the quasi steady state rate as well as a slower increase and decrease after start and end of the oxygen exposure, respectively, show a kinetically relevant contribution of methyl formate to the observed water formation. The reaction probability of oxygen towards water can be estimated to be lower by a factor of approx. 60 for methyl formate than for methanol considering the quasi-steady-state rate during the pulse is halved (Fig. 2a and b) and the flux is increased by a factor of approx. 30 for methyl formate (combined with quantitative oxygen consumption by the end of the delay time). This strong reactivity difference suggests gold surfaces allowing for high methanol conversions, before overoxidation of the partial oxidation product methyl formate significantly contributes to a selectivity loss, as desired for an ideal partial oxidation catalyst.
But how can the reduced reactivity of oxygen towards methyl formate be understood given the fact that oxidation was observed as low as 125 K18 indicative of a low activation barrier? As the desorption temperatures of methyl formate and methanol from bare Au(332) are very similar (see Fig. S3 (ESI†) and ref. 18), it is expected that the ratio of their surface concentrations is similar to the ratio of the applied fluxes in the gas phase. Thus, the lower reactivity cannot be explained by a significantly reduced availability of methyl formate on the surface. In this regard, it is important to recall that methyl formate oxidation occurs only, with a subset of the oxygen atoms present at low coverage which are subsumed as Olc,sp in Fig. 3. Additionally, the three reaction channels for the overoxidation of methyl formate proceed along different reaction pathways suggesting rather specific transition state geometries.18 These requirements render the probability of successful reaction events low. The slow transient kinetics of the water production show that methyl formate does not react rapidly with oxygen atoms (O, Fig. 3) which impinge on the surface and are transiently present in these experiments. The slow reactivity of methyl formate with oxygen leads to an accumulation of unreacted oxygen during the pulse preferentially accommodating at low coordinated sites (Olc, Fig. 3). A subset of these were shown to allow for methyl formate oxidation (Olc,sp) resulting in an increase of the rate during the oxygen pulse and leading to a constant water formation rate as soon as the reactive surface sites are populated with oxygen.
 | ||
| Fig. 3 Schematic representation of reactions of oxygen on Au(332) with methanol and methyl formate.18,20,21 Olc denoting oxygen on low-coordinated sites, Olc,sp the subset of Olc active in the methyl formate oxidation, O all other types of oxygen species on the Au(332) surface and P denoting other products. Double-crossed reaction arrows indicate that the respective reaction does not occur at a significant rate. | ||
In contrast to that, partial oxidation of methanol is fast and the transient kinetics shows that the oxygen concentration during the pulses is very small. This is in line with a previous MB study on the partial oxidation of methanol which indicates an effective reaction of methanol with different types of oxygen species, i.e. without specific requirements for oxygen to be located on specific surface sites.20 Hence, methanol reacts with all oxygen species on the surface as indicated in Fig. 3.
As methanol was found to react so much faster than methyl formate, it is at first glance surprising that co-feeding methyl formate together with methanol has a significant impact on the reactivity of the system. The slower rate increase of water formation for the co-feed as compared to pure methanol indicates that methyl formate can alter the availability of oxygen for the reaction with methanol at least at the beginning of the pulse. This behaviour can be understood by methyl formate interaction with activated oxygen on the surface, denoted as [O⋯H3COCHO] in Fig. 3, reducing the probability of a reactive encounter with transiently adsorbed methanol (Fig. 3). However, these transient [O⋯H3COCHO] species will dissociate without reaction in most of the cases (Fig. 3). In this respect, it is also important to recall that the methyl formate flux is about 30 times that of methanol which is also the ratio for collisions between oxygen and the molecules in case of similar residence times (s.a.). The ability of methyl formate to interact with activated oxygen was already demonstrated by TPD showing an increase of the desorption temperature of molecular methyl formate desorption by about 15 K, if comparing oxygen pre-covered with the pristine Au(332) surface.18 A reduction of the reaction rate will result in an accumulation of oxygen on the surface allowing for an increase of methanol oxidation at later times during the pulse which will surpass the reaction rate of methyl formate because of the reduced requirements of methanol with respect to the nature of the oxygen species.
Np-Au is expected to contain also specific low-coordinated sites active in methyl formate overoxidation. However, this subset of low-coordinated sites is presumably also a minority species on np-Au catalysts exhibiting a high density of low-coordinated sites, such as steps and kinks, in curved regions of the ligaments, but also [111]- and [100]-facets in flat regions.9 Under typical isothermal conditions with very low oxygen coverages (saturation coverage of approx. 0.004 ML11,12), these overoxidation channels will not be kinetically important on np-Au until high conversions are reached. Yet, at high conversion formation of [O⋯H3COCHO] species may slow methanol oxidation.
In conclusion, we have shown that low barrier pathways for the oxidation of methyl formate are available on stepped Au(332) surfaces at low oxygen coverage. However, the overoxidation of methyl formate under isothermal oxygen lean conditions is slow as compared to the oxidation of methanol at the same temperature and oxygen fluxes. This is due to fact that special oxygen species being minority species on the surface are required for the overoxidation of methyl formate whereas methanol is less selective and reacts with various oxygen species. Hence, the heterogeneous reactivity of different oxygen species on gold surfaces is critical for the high selectivity in the aerobic partial oxidation of methanol towards methyl formate. These results not only emphasise the potential of np-Au catalysts, but importantly provide an improved microscopic understanding required for a rational improvement of these catalysts.
Conflicts of interest
There are no conflicts to declare.Notes and references
- D. Kaiser, L. Beckmann, J. Walter and M. Bertau, Catalysts, 2021, 11, 869 CrossRef CAS.
- L. Rong, Z. Xu, J. Sun and G. Guo, J. Energy Chem., 2018, 27, 238–242 CrossRef.
- P. Klezl, EU Pat., EP0501097, 1995 Search PubMed.
- J. S. Lee, J. C. Kim and Y. G. Kim, Appl. Catal., 1990, 57, 1–30 CrossRef CAS.
- S. Jali, H. B. Friedrich and G. R. Jullius, J. Mol. Catal. A: Chem., 2011, 348, 63–69 CrossRef CAS.
- A. Wittstock, V. Zielasek, J. Biener, C. M. Friend and M. Bäumer, Science, 2010, 327, 319–322 CrossRef CAS PubMed.
- J. Erlebacher, M. J. Aziz, A. Karma, N. Dimitrov and K. Sieradzki, Nature, 2001, 410, 450–453 CrossRef CAS PubMed.
- L. C. Wang, Y. Zhong, D. Widmann, J. Weissmüller and R. J. Behm, ChemCatChem, 2012, 4, 251–259 CrossRef CAS.
- T. Fujita, P. F. Guan, K. McKenna, X. Y. Lang, A. Hirata, L. Zhang, T. Tokunaga, S. Arai, Y. Yamamoto, N. Tanaka, Y. Ishikawa, N. Asao, Y. Yamamoto, J. Erlebacher and M. W. Chen, Nat. Mater., 2012, 11, 775–780 CrossRef CAS PubMed.
- L. C. Wang, Y. Zhong, H. J. Jin, D. Widmann, J. Weissmüller and R. J. Behm, Beilstein J. Nanotechnol., 2013, 4, 111–128 CrossRef PubMed.
- L. C. Wang, M. L. Personick, S. Karakalos, R. Fushimi, C. M. Friend and R. J. Madix, J. Catal., 2016, 344, 778–783 CrossRef CAS.
- L. C. Wang, C. M. Friend, R. Fushimi and R. J. Madix, Faraday Discuss., 2016, 188, 57–67 RSC.
- B. Zugic, L. C. Wang, C. Heine, D. N. Zakharov, B. A. J. Lechner, E. A. Stach, J. Biener, M. Salmeron, R. J. Madix and C. M. Friend, Nat. Mater., 2017, 16, 558–564 CrossRef CAS PubMed.
- B. J. Xu, X. Y. Liu, J. Haubrich, R. J. Madix and C. M. Friend, Angew. Chem., Int. Ed., 2009, 48, 4206–4209 CrossRef CAS PubMed.
- J. J. Pireaux, M. Chtaib, J. P. Delrue, P. A. Thiry, M. Liehr and R. Caudano, Surf. Sci., 1984, 141, 211–220 CrossRef CAS.
- A. G. Sault, R. J. Madix and C. T. Campbell, Surf. Sci., 1986, 169, 347–356 CrossRef CAS.
- M. J. Prieto, E. A. Carbonio, R. Landers and A. de Siervo, Surf. Sci., 2013, 617, 87–93 CrossRef CAS.
- C. D. Feldt, J. L. Low, P. A. Albrecht, K. Tang, W. Riedel and T. Risse, J. Phys. Chem. C, 2021, 23, 21599–21605 CAS.
- Y. Li, W. Dononelli, R. Moreira, T. Risse, M. Bäumer, T. Klüner and L. V. Moskaleva, J. Phys. Chem. C, 2018, 122, 5349–5357 CrossRef CAS.
- C. D. Feldt, T. Gimm, R. Moreira, W. Riedel and T. Risse, Phys. Chem. Chem. Phys., 2021, 23, 21599–21605 RSC.
- C. D. Feldt, T. Kirschbaum, J. L. Low, W. Riedel and T. Risse, Catal. Sci. Technol., 2022, 12, 1418–1428 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details and supporting Figures. See DOI: 10.1039/d1cc06939j |
| This journal is © The Royal Society of Chemistry 2022 |