
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Helicity control of a perfluorinated carbon chain within a chiral supramolecular cage monitored by VCD†
Carlo
Bravin
 a,
Giuseppe
Mazzeo
b,
Sergio
Abbate
b,
Giulia
Licini
a,
Giovanna
Longhi
*b and
Cristiano
Zonta
*a
a,
Giuseppe
Mazzeo
b,
Sergio
Abbate
b,
Giulia
Licini
a,
Giovanna
Longhi
*b and
Cristiano
Zonta
*a
aDipartimento di Scienze Chimiche, Università degli Studi di Padova, Padova, PD, Italy. E-mail: cristiano.zonta@unipd.it
bDepartment of Molecular and Translational Medicine, Università di Brescia, Viale Europa 11, 25123 Brescia, BS, Italy. E-mail: giovanna.longhi@unibs.it
First published on 14th January 2022
Abstract
Confinement within supramolecular systems is the leading technology to finely tune guest functional properties. In this communication we report the synthesis of a chiral supramolecular cage able to bias the helicity of a perfluorinated carbon chain hosted within the cage. We monitor the phenomenon of chiral induction by Vibrational Circular Dichroism (VCD) experiments complemented by DFT calculations over the possible conformers.
Helicity represents a major chiral motif found in natural and artificial systems.1 It is manifested at the macro- and supramolecular level in a large number of structures, among which the α-helix in the secondary structure of proteins and the double helix formed by two strands of DNA are the most prominent examples. Interestingly, perfluorinated alkane chains longer than four carbon atoms also adopt helical conformations.2 In contrast to the all-anti conformation of corresponding perhydroalkanes, the twisted nature of the perfluorinated carbon chain is due to hyperconjugative stabilization through σCC–σ*CF interaction.3 This particular feature has been observed in the solid state as well as in solution.4 In particular, from the solid state analysis, a full 360° twist occurs in 33.6 Å (13 zigzags or 26 chain atoms) and the angle between F1 and F5 respectively bound to C1 and C3 (projected 1–5 F⋯F angle) is 27°. This helical conformation results in two enantiomeric helices.
In recent years, due to the interest in the development of functional materials with defined helicity in the structure, stereo-induction phenomena onto perfluoro-chains were investigated. In particular, covalent attachment of groups endowed with a stereogenic centre was exploited in order to bias their helicity.5 A relevant example, in which the presence of a stereogenic centre induces preferential helical chirality, was reported by Monde et al.5a In particular, the covalent linkage of a chiral benzyl alcohol to a perfluorinated chain resulted in a preferential induction of the helical chain handedness. To confirm the preferential formation of one helix in solution, the Vibrational Circular Dichroism (VCD) technique was successfully applied to reveal the helix absolute configuration.
Inspired by this study, we wondered if it were possible to induce chirality in a perfluoroalkyl chain using non-covalent interactions arising from encapsulation of a perfluoroalkyl chain in a chiral confined space.6 It should be highlighted that Ajami and Rebek already reported that the incorporation of an alkyl chain within a supramolecular capsule drove the alkyl chain from the fully extended conformation to a coiled, compressed conformation.7 While this capsule exerted the ability to “coil” the guest present in the interior of the cavity, the possibility to control the twist direction of the guest chain still represents a challenge for a confined system.
In this article we report the synthesis of a chiral cage based on tris(2-pyridylmethyl)amine TPMA8 metal complexes,9 and the effect that it can induce over an included perfluorinated dicarboxylic acid. The helix configuration of the perfluoroalkyl diacidic chain was evaluated with a combination of VCD experiments and theoretical DFT calculations of the host–guest complex. VCD experiments, in fact, revealed to be crucial in determination of the conformation of the supramolecular inclusion complex in solution.10
The chiral supramolecular cage with the included perfluoro diacid was synthesized adapting a synthetic strategy previously used for the corresponding perhydrocarbon system.9e In particular, slow addition of the chiral diamine linker (1R,2R)-cyclohexanediamine (2.5 equiv.) to a dilute DMSO-d6 solution of initial Zn(II) complex (1.0 equiv.) in the presence of a octafluoroadipic acid C6F (1.0 equiv.) led to included cage C6F@(R,R)-1 (Scheme 1 and Section S2.1, ESI†). Formation of the system was monitored by 1H NMR. In particular, aldehyde proton signal at 10.03 ppm disappears in favour of the formation of the imine signal at 8.49 ppm (Fig. S3.1A, ESI†). The presence of the cage in solution was confirmed also by 2D NMR experiments COSY and DOSY (Fig. S3.1B and C, ESI†), and ESI-MS analysis. In the latter, formation of inclusion system C6F@(R,R)-1 was confirmed by molecular ion signals at 929.2 m/z with the correct isotopic pattern (Fig. S3.3, ESI†). IR spectra of C6F@(R,R)-1 were recorded in DMSO-d6 revealing the diagnostic signals related to the guest, in particular the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching (1690 cm−1), carboxylate symmetric stretching (1360 cm−1)11 and the C–F stretching 1170 cm−1, however the overall intensity was too low (∼0.2–0.1 A) to record a VCD spectrum. It should be noted that VCD is approximately 4 to 6 orders of magnitude weaker than IR and for this reason the synthesis had to be optimized to ensure the inclusion cage compound at suitable concentrations.
O stretching (1690 cm−1), carboxylate symmetric stretching (1360 cm−1)11 and the C–F stretching 1170 cm−1, however the overall intensity was too low (∼0.2–0.1 A) to record a VCD spectrum. It should be noted that VCD is approximately 4 to 6 orders of magnitude weaker than IR and for this reason the synthesis had to be optimized to ensure the inclusion cage compound at suitable concentrations.
 | ||
| Scheme 1 C6F@(R,R)-1 cage is obtained by imine condensation reaction between a tris-aldehyde metal-complex, (R,R)-dicyclohexylamine and octafluoroadipic C6F diacid as templating agent. | ||
Different samples of C6F@(R,R)-1 at increasing cage concentrations were prepared only by modifying the concentration of initial components. The cage samples were synthetized at 5, 10 and 15 mM final concentration (Fig. S3.1D, ESI†). Suitable absorbance for the VCD analysis, namely values between 0.2–0.9 A, were obtained for the sample containing the cage at 10 mM.
VCD spectroscopy, after the first pioneering works in the seventies,12 is currently a reliable method to assign the absolute configuration (AC) of organic molecules13 and is in principle generally applicable, since it does not need the presence of UV-VIS chromophores like Electronic CD (ECD).14 Although the VCD technique has several limitations, in particular disadvantages related to weaker signals, the main advantage of this technique that makes it an essential tool for determining the absolute configuration of chiral molecules is the straightforward simulation the VCD spectrum for each stereoisomer that can be compared with the experimental one usually relying on several signals.15 Besides that, the technique is quite sensitive to molecular conformations and intermolecular/supramolecular interactions,10,16 such that a good correspondence between calculations and experiments may confirm the picture obtained by the theoretical modelling.
VCD spectra of both enantiomers C6F@(R,R)-1 and C6F@(S,S)-1 have been recorded in 10 mM DMSO solution (Fig. S6.1, ESI†), the semi-difference spectrum is reported in Fig. 1. The most evident signal is a couplet centred at about 1370 cm−1, at higher wavenumber no clear VCD feature has been observed apart from a weak couplet in the carbonyl-stretching region; in the range 1300–1150 cm−1 weaker and broader VCD features are observed. DMSO-d6 does not permit to record VCD signals at lower wavenumber: thus, the only typical CF stretching features which we were able to observe are at about 1170 cm−1.
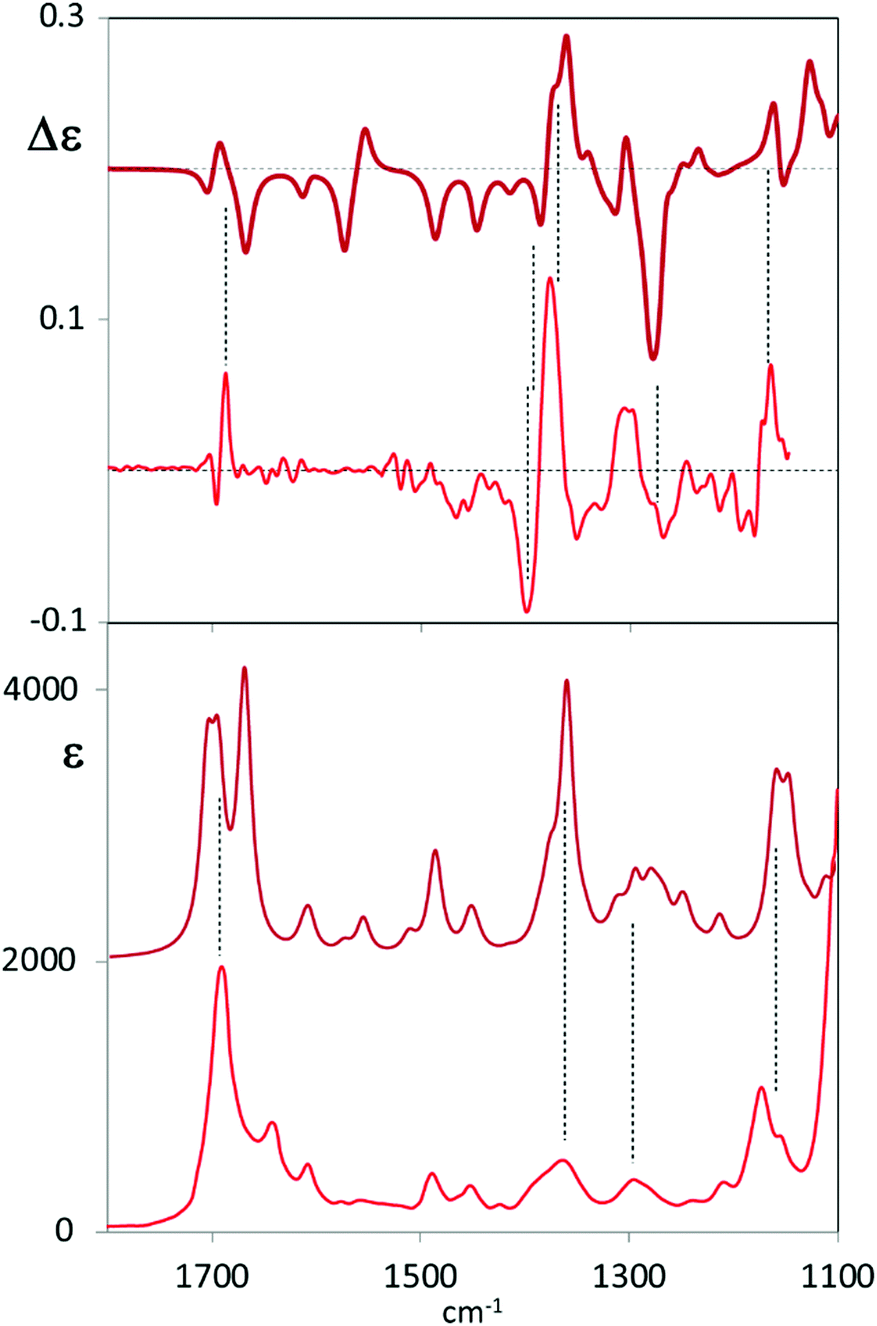 | ||
| Fig. 1 Comparison of experimental (lower traces) VCD spectra of C6F@(R,R)-1 (top square) and IR absorption spectra (lower square) of (R,R)-C6F@2 (see Scheme 1) with calculated (upper traces) VCD and IR spectra of C6F@(R,R)-1 with the two head groups in P,P configuration and with the P configuration of the fluorinated chain. Experimental data are displayed with weaker lines in the lower part of each panel; main correspondences are evidenced with vertical lines. | ||
To gain structural information from VCD data, comparison with calculated spectra is necessary. Due to the complexity of the system under study, conformational analysis for C6F@(R,R)-1 was undertaken and all possible diastereoisomers were considered on the basis of the stereochemical elements present in the system. As a consequence of the propeller-like arrangement of the ligand around the metal,17 the empty cage can exhibit three main diastereoisomeric conformations: two cages with D3 symmetry in which the two helical arrangements of TPMA are the same (MM, PP) and one in which the two helical arrangements are opposite (MP). Within these cages the perfluorinated carbon chain can adopt either P or M helical forms providing thus a total of six diastereoisomers: MM–M, MM–P, MP–M, MP–P, PP–M, PP–P (Section S4, ESI†). DFT B3LYP/6-31G(d,p) calculations were run with Gaussian 16 package Revision B.0118 starting from six different structures manually generated. Calculated relative energies revealed that the most stable conformation of the inclusion cage is PP–P (Fig. 2 and Table S4.1, ESI†). Inversion of the internal perfluorinated carbon chain (viz. PP–M) results in a structure which is 1 kcal mol−1 energy less stable; even higher is the cost in the four remaining cases, where at least one TPMA moiety is present in the M configuration. Nonetheless, optimized DFT structures are quite regular and respect the proposed classification, see for example the NCCN dihedral angles reported in (Table S4.1, ESI†). Other dihedral angles that may influence the chiroptical response are the ones between phenyl and pyridine groups, that are all found to be regularly oriented (either M or P depending on the cage).
 | ||
| Fig. 2 DFT minimised structure of C6F@(R,R)-1. | ||
The perfluorinated diacidic chain is always elongated, tilted from trans-planar conformation as expected.4 However, to hinge the carboxylic groups to the Zn ions of the cage, the chain is slightly more elongated and more trans-planar than a corresponding chain not entrapped in the cage (Table S4.1, last column, ESI†). The helicity of the perfluorinated chain is related also to the mutual orientation of the two terminal COO groups which is indicated by dihedral angles (Table S4.1, Fig. S4.1 and ESI†).
From Fig. 1 one may appreciate the acceptable correspondence between the experimental VCD spectrum and the one calculated for the PP–P geometry, supporting the idea that in DMSO solution this is the most stable structure. In any case the VCD and IR spectra of each diastereoisomer were calculated and are presented in Fig. 3. The comparison of the six calculated spectra is quite instructive and shows that the (+,−) doublet in order of increasing energy centred at 1370 cm−1 is associated to P orientation of the two carboxylate groups (and correspondingly to P helicity of the perfluorinated chain) while the (−, +) doublet is related to the M orientation, irrespective of cage chirality. On the contrary, the features calculated in the 1300–1200 cm−1 and 1450–1600 cm−1 ranges correlate to cage chirality. Some calculated wavenumbers and rotational strengths are reported in Table S2 (ESI†) for a normal mode analysis.
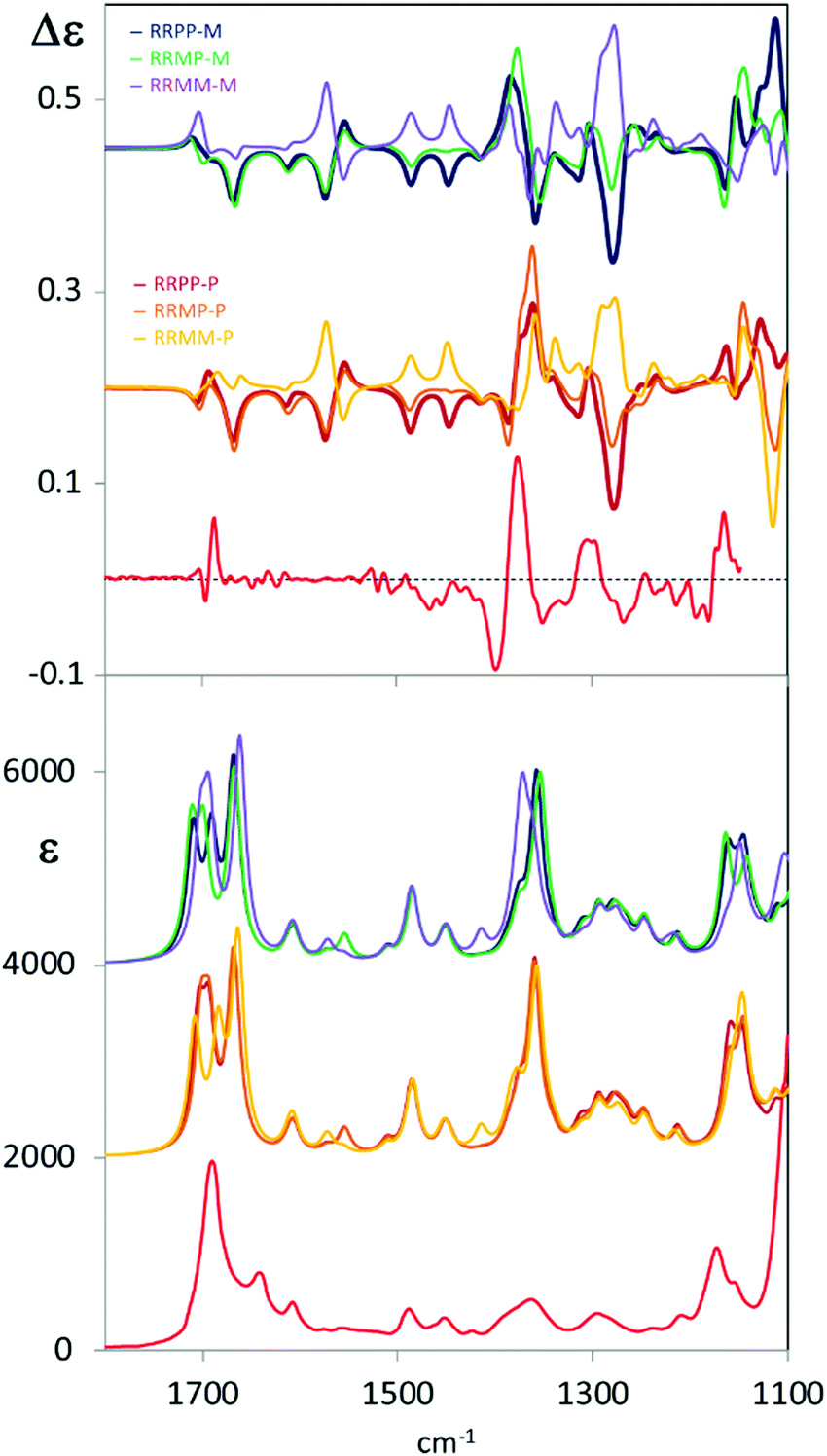 | ||
| Fig. 3 Comparison of experimental VCD spectra of C6F@(R,R)-1 (top) and IR absorption spectra of C6F@(R,R)-1 with calculated VCD and IR spectra of C6F@(R,R)-1 with the two head groups and fluorinated chains in all possible configurations (PP-M, PP-P, MM-M, MM-P, MP-M, MP-P). Experimental data in red. | ||
One may associate the intense doublet at 1370 cm−1 to carboxylate symmetric stretchings: the high energy component is attributed to the in phase vibrational modes in the two moieties; the low energy component receives contributions from out of phase symmetric carboxylate stretchings (PP–P, MP–P, MP–M and MM–M) while for MM–P and PP–M the low energy component of the doublet receives contributions prevalently from vibrations localized on the cage. The CF stretching bands at 1100–1190 cm−1 are less diagnostic for two main reasons: they are located in the lowest accessible spectroscopic range5,19 and also aromatic CH bendings of the cage and CH2 vibrations of the chiral cyclohexane ring are present there. Overall, considering observed versus calculated VCD features, we may notice that the doublet attributed to the guest appears quite intense and sharp as suggested by calculations, on the contrary, VCD bands due to vibrations of the cage, particularly the ones associated to the lateral chains, are broader and weaker. Possible perturbations due to DMSO-d6 molecules have not been considered in the calculations. It is interesting to notice that in the high energy region we are not able to detect bands associated to cage vibrational modes, that is the in plane bending of the CH adjacent to the cyclohexyldiamine (band calculated at 1720 cm−1), or the in plane bendings of the phenyl–pyridyl groups; on the contrary we can detect the carbonyl doublet at 1700 cm−1 (calculated at 1750 cm−1). Such observations and the consideration of similarity index (0.42 for PP–P, 0.61 for MP–P, 0.55 for an average of PP–P and MM–P) might suggest that we are observing the meso form MP of the cage or that both the MM–P and PP–P forms are present. Since DFT-predicted conformational preferences obtained for the isolated, implicitly solvated molecule may not provide the best representation of the spectroscopic response of a molecule in solution, it is tempting to use VCD to determine the real conformer populations.20 However, another possible explanation could be that the external groups of the cage are more perturbed by the solvent, thus exhibiting high conformational flexibility, such that averaging over many structures could give weak VCD signals.21 On the contrary, vibrational modes of the guest-molecule and of tris(2-pyridylmethyl)amine rigidified by Zn complexation (1300–1200 cm−1) are much more evident. As a final note, with the two carbonyls of the carboxylate groups showing 1.23 Å calculated length for the free CO and 1.27 Å for the one bound to Zn2+, the antisymmetric stretching is more localized on the free CO (1750 cm−1), while the symmetric one is delocalized to the same extent on the two CO bonds (1402 and 1428 cm−1). Major importance for our conclusions has the experimental ECD spectra in comparison to the ones calculated for the six conformational diastereoisomers (Fig. S6.2 and S7.1, ESI†). ECD in the accessible region is sensitive only to the cage configuration/conformation confirming the PP structure.
In conclusion, a chiral molecular cage able to control the helicity of an octafluoroadipic dicarboxylic acid embedded within its cavity is reported.22 The reaction conditions were optimized to obtain a suitable instrumental response of the VCD analysis, and computational studies were carried out to identify the most stable diastereoisomeric cage in solution. VCD and absorption spectra of C6F@(R,R)-1 are well predicted by the calculations and are in line with experimental observations. These results confirmed the capability of the cage to induce a preferential helicity of the perfluorinated guest.
Department of Chemical Sciences at University of Padova is (P-DiSC#10 BIRD2020-UNIPD, LICC); Big&Open Data Innovation Laboratory (BODaI-Lab); University of Brescia; Computing Center CINECA (Bologna, Italy); Italian Ministry of University and Research (PRIN 2017 program 2017A4XRCA_003).
Conflicts of interest
There are no conflicts to declare.Notes and references
- (a) Y. Wang, J. Xu, Y. Wanga and H. Chen, Chem. Soc. Rev., 2013, 42, 2930–2962 RSC; (b) E. Yashima, N. Ousaka, D. Taura, K. Shimomura, T. Ikai and K. Maeda, Chem. Rev., 2016, 116, 13752 CrossRef CAS PubMed; (c) B. A. F. Le Bailly and J. Clayden, Chem. Commun., 2016, 52, 4852 RSC.
- C. W. Bunn and E. R. Howells, Nature, 1954, 174, 549 CrossRef CAS.
- R. A. Cormanich, D. O'Hagan and M. Bühl, Angew. Chem., Int. Ed., 2017, 56, 7867 CrossRef CAS PubMed.
- (a) K. Ute, R. Kinoshita, K.-I. Matsui, N. Miyatake and K. Hatada, Chem. Lett., 1992, 1337 CrossRef CAS; (b) S. S. Jang, M. Blanco, W. A. Goddard, G. Caldwell and R. B. Ross, Macromolecules, 2003, 36, 5331 CrossRef CAS; (c) C. Quarti, A. Milani and C. Castiglioni, J. Phys. Chem. B, 2013, 117(2), 706 CrossRef CAS PubMed; (d) T. Hasegawa, T. Shimoaka, N. Shioya, K. Morita, M. Sonoyama, T. Takagi and T. Kanamori, ChemPlusChem, 2014, 79, 1421 CrossRef CAS.
- (a) K. Monde, N. Miura, M. Hashimoto, T. Taniguchi and T. Inabe, J. Am. Chem. Soc., 2006, 128, 6000 CrossRef CAS PubMed; (b) H. Sato, T. Yajima and A. Yamagishi, Chirality, 2016, 28, 361 CrossRef CAS PubMed; (c) H. Sato, T. Yajima and A. Yamagishi, Chem. Commun., 2011, 47, 3736 RSC; (d) K. Kohno, K. Morimoto, N. Manabe, T. Yajima, A. Yamagishi and H. Sato, Chem. Commun., 2012, 48, 3860 RSC; (e) L. Zhang, S. Lin, Q. Tong, Y. Li, Y. Wang, Y. Li, B. Li and Y. Yang, Chirality, 2019, 31, 992 CrossRef CAS PubMed; (f) L. Zhang, S. Lin, Y. Li, B. Lia and Y. Yang, New J. Chem., 2021, 45, 3193 RSC.
- (a) G. Qiu, C. Colomban, N. Vanthuyne, M. Giorgi and A. Martinez, Chem. Commun., 2019, 55, 14158 RSC; (b) P. Qiu, P. Nava, C. Colomban and A. Martinez, Front. Chem., 2020, 8, 994 Search PubMed; (c) A. Martinez, L. Guy and J.-P. Dutasta, J. Am. Chem. Soc., 2010, 132, 16733 CrossRef CAS PubMed; (d) D. Zhang, T. K. Ronson, J. L. Greenfield, T. Brotin, P. Berthault, E. Léonce, J.-L. Zhu, L. Xu and J. R. Nitschke, J. Am. Chem. Soc., 2019, 141, 8339 CrossRef CAS PubMed.
- D. Ajami and J. Rebek, Nat. Chem., 2009, 1, 87 CrossRef CAS PubMed.
- C. Bravin, E. Badetti, G. Licini and C. Zonta, Coord. Chem. Rev., 2021, 427, 213558 CrossRef CAS.
- (a) C. Bravin, E. Badetti, F. A. Scaramuzzo, G. Licini and C. Zonta, J. Am. Chem. Soc., 2017, 139, 6456 CrossRef CAS PubMed; (b) C. Bravin, E. Badetti, R. Puttreddy, F. Pan, K. Rissanen, G. Licini and C. Zonta, Chem. – Eur. J., 2018, 24, 2936 CrossRef CAS PubMed; (c) F. Begato, R. Penasa, G. Licini and C. Zonta, Chem. Commun., 2021, 57, 10019 RSC; (d) C. Bravin, J. A. Piękoś, G. Licini, C. A. Hunter and C. Zonta, Angew. Chem., Int. Ed., 2021, 60, 23871 CrossRef CAS PubMed; (e) C. Bravin, G. Mason, G. Licini and C. Zonta, J. Am. Chem. Soc., 2019, 141, 11963 CrossRef CAS PubMed; (f) C. Bravin, A. Guidetti, G. Licini and C. Zonta, Chem. Sci., 2019, 10, 3523 RSC.
- (a) V. Setnicka, M. Urbanova, K. Volka, S. Nampally and J.-M. Lehn, Chem. – Eur. J., 2006, 12, 8735 CrossRef CAS PubMed; (b) P. Zhang and P. L. Polavarapu, J. Phys. Chem. A, 2007, 111(5), 858 CrossRef CAS PubMed; (c) S. Abbate, G. Longhi, A. Ruggirello and V. Turco Liveri, Colloids Surf., A, 2008, 327, 44 CrossRef CAS; (d) H. Sato, Phys. Chem. Chem. Phys., 2020, 22, 7671 RSC.
- (a) C. C. R. Sutton, G. da Silva and G. V. Franks, Chem. – Eur. J., 2015, 21, 6801 CrossRef CAS PubMed; (b) M. Justi, M. Puggina de Freitas, J. M. Silla, C. A. Nunes and C. A. Silva, J. Mol. Struct., 2021, 130405 CrossRef CAS.
- (a) G. Holzwarth, H. S. Mosher, T. R. Faulkner and A. Moscowitz, J. Am. Chem. Soc., 1974, 96, 251 CrossRef CAS; (b) L. A. Nafie, T. A. Keiderling and P. J. Stephens, J. Am. Chem. Soc., 1976, 98, 2715 CrossRef CAS.
- (a) L. A. Nafie, Vibrational Optical Activity: Principles and Applications, John Wiley & Sons, Ltd, New York, 2011 CrossRef; (b) P. L. Polavarapu, Chirality, 2012, 24, 909 CrossRef CAS PubMed.
- G. Pescitelli, L. Di Bari and N. Berova, Chem. Soc. Rev., 2014, 43, 5211 RSC.
- (a) M. Fuse, G. Mazzeo, G. Longhi, S. Abbate, D. Zerla, I. Rimoldi, A. Contini and E. Cesarotti, Chem. Commun., 2015, 51, 9385 RSC; (b) G. Mazzeo, A. Cimmino, M. Masi, G. Longhi, L. Maddau, M. Memo, A. Evidente and S. Abbate, J. Nat. Prod., 2017, 80, 2406 CrossRef CAS PubMed; (c) L. Paoloni, G. Mazzeo, G. Longhi, S. Abbate, M. Fusè, J. Bloino and V. Barone, J. Phys. Chem. A, 2020, 124(5), 1011 CrossRef CAS PubMed; (d) C. Merten, J. Bloino, V. Barone and Y. Xu, J. Phys. Chem. Lett., 2013, 4, 3424 CrossRef CAS.
- (a) C. Merten and Y. Xu, Angew. Chem., Int. Ed., 2013, 52, 273 Search PubMed; (b) P. Reiné, A. M. Ortuño, S. Resa, L. Álvarez de Cienfuegos, V. Blanco, M. J. Ruedas-Rama, G. Mazzeo, S. Abbate, A. Lucotti, M. Tommasini, S. Guisán-Ceinos, M. Ribagorda, A. G. Campaña, A. Mota, G. Longhi, D. Miguel and J. M. Cuerva, Chem. Commun., 2018, 54, 13985 RSC; (c) S. Ghidinelli, S. Abbate, G. Mazzeo, S. Boiadjiev, D. A. Lightner and G. Longhi, Phys. Chem. Chem. Phys., 2021, 23, 20138 RSC.
- C.-L. Chuang, K. Lim, Q. Chen, J. Zubieta and J. W. Canary, Inorg. Chem., 1995, 34, 2562 CrossRef CAS.
- M. J. Frisch, et al., Gaussian 16, Revision B.01, Gaussian, Inc., Wallingford CT, 2016 Search PubMed.
- S. Abbate, G. Longhi, G. Mazzeo, C. Villani, S. Petković and R. Ruzziconi, RSC Adv., 2019, 9, 11781 RSC.
- (a) M. A. J. Koenis, Y. Xia, S. R. Domingos, L. Visscher, W. J. Buma and V. P. Nicu, Chem. Sci., 2019, 10, 7680 RSC; (b) M. Kemper, E. Engelage and C. Merten, Angew. Chem., Int. Ed., 2021, 60, 2958 CrossRef CAS PubMed.
- S. Ghidinelli, S. Abbate, J. Koshoubu, Y. Araki, T. Wada and G. Longhi, J. Phys. Chem. B, 2020, 124, 4512 CrossRef CAS PubMed.
- Preliminary investigations of the corresponding perhydro diacid are reported in the ESI†.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details and characterization of all new compounds; selected 2D-NMR experiments (COSY, DOSY) and the detailed conformational search among the structures (DFT, TD-DFT calculations). See DOI: 10.1039/d1cc06861j |
| This journal is © The Royal Society of Chemistry 2022 |