
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Targeted delivery of maytansine to liver cancer cells via galactose-modified supramolecular two-dimensional glycomaterial†
Hai-Na
Xie‡
b,
Yu-Yuan
Chen‡
a,
Guo-Biao
Zhu
a,
Hai-Hao
Han
 *a,
Xi-Le
Hu
a,
Zhi-Qiang
Pan
b,
Yi
Zang
c,
Dong-Hao
Xie
*d,
Xiao-Peng
He
a,
Jia
Li
*c and
Tony D.
James
*ef
*a,
Xi-Le
Hu
a,
Zhi-Qiang
Pan
b,
Yi
Zang
c,
Dong-Hao
Xie
*d,
Xiao-Peng
He
a,
Jia
Li
*c and
Tony D.
James
*ef
aKey Laboratory for Advanced Materials and Joint International Research Laboratory of Precision Chemistry and Molecular Engineering, Feringa Nobel Prize Scientist Joint Research Center, Frontiers Center for Materiobiology and Dynamic Chemistry, School of Chemistry and Molecular Engineering, East China University of Science and Technology, 130 Meilong Rd., Shanghai 200237, P. R. China. E-mail: hanhaihao@126.com
bSchool of Basic Medicinal Sciences, Shanghai University of Traditional Chinese Medicine, No. 1200 Cailun Rd., Shanghai, P. R. China
cNational Center for Drug Screening, State Key Laboratory of Drug Research, Shanghai Institute of Materia Medica, Chinese Academy of Sciences, 189 Guo Shoujing Rd., Shanghai 201203, P. R. China. E-mail: jli@simm.ac.cn
dDepartment of Pharmacy, Shanghai Guanghua Hospital of Integrative Medicine, No. 540 Xinghua Rd., Shanghai, P. R. China. E-mail: xdhdh2012@126.com
eDepartment of Chemistry, University of Bath, Bath, BA2 7AY, UK. E-mail: t.d.james@bath.ac.uk
fSchool of Chemistry and Chemical Engineering, Henan Normal University, Xinxiang 453007, China
First published on 19th March 2022
Abstract
A two-dimensional (2D) glycomaterial for targeted delivery of maytansine to liver cancer cells was developed. Host–guest interaction between a galactosyl dye and human serum albumin (HSA) produces supramolecular galactoside–HSA conjugates, which are then used to coat 2D MoS2. The 2D glycomaterial was shown to be capable of the targeted delivery of maytansine to a liver cancer cell line that highly expresses a galactose receptor, resulting in greater cytotoxicity than maytansine alone.
Liver cancer is amongst one of the most common malignant tumours. According to the International Agency for Research on Cancer (IARC) World Cancer Report 2020, liver cancer results in the third highest mortality rate for all types of tumours with a five-year survival rate of less than 18%. This poses a serious threat to human health and the global economy.1 Therefore, the development of effective therapeutic methods is crucial to improve the prognosis and improve the survival rate of patients diagnosed with liver cancer.
Despite rapid progress in the development of antibody- and cell-based therapeutics, chemotherapy, based on cytotoxic drugs remains the major method for liver cancer treatment in hospitals worldwide.2 Conventional chemotherapeutic agents fail to distinguish between normal and cancer cells, thereby leading to serious side effects and an overall low therapeutic efficacy. Recent research has identified multiple key biomarkers (e.g. glypican-3)3,4 in the development and progression of liver cancer, facilitating the development of targeted therapeutics (e.g. antibody–drug conjugates), ADCs, and Chimeric Antigen Receptor T-Cell Immunotherapy, CAR-T, to selectively kill cancer cells without affecting the growth of normal cells.5 However, ADCs are costly to prepare due to the complexity in the production process of small molecule-biomacromolecule conjugates. This is also the problem for preparing CAR-T based therapeutic agents. Moreover, ADCs and CAR-T are much more expensive when compared to pure chemotherapeutic drugs, and as such impose a large economic burden on patients.
Carbohydrate–protein interactions (CPIs) are one of the most ubiquitous biological events on the surface of cell membranes, and play an important role in modulating biological processes.6 The development of drug delivery strategies through the covalent or non-covalent attachment of carbohydrates to a variety of bioactive compounds and material substrates has proven effective in the field of drug-targeted cancer therapy. For example, Liu et al. reported glucosylated triptolide, which allows for targeted drug release in cancer cells with sustained antitumour activity.7 More recently, three glycosylated small-interfering RNA (siRNA)-based therapeutic agents have already been approved for clinical use by the US Food and Drug Administration (FDA) and European Medicines Agency (EMA).8 Given these promising examples, we believe carbohydrate-modified therapeutic agents could become a promising alternative to antibody and cell-based therapies. However, effort in this field has been limited to the covalent modification of therapeutic agents with carbohydrates, whereas the exploitation of supramolecular strategies remain less explored. We9 and others10 have recently reported that HSA, which is the serum protein abundant in the human body, is a suitable supramolecular host for accommodating a diverse range of fluorescent diagnostic and theranostic probes with enhanced properties. This offers scope for the construction of supramolecular “glycoproteins” for biomedical applications through the simple inclusion of a dye-modified glycoside into the hydrophobic cavity of HSA.9,11a
Multivalency is an important factor for carbohydrates to achieve high avidity with their binding proteins.12,13 While previous studies focused on the covalent grafting of multiple glycosyl units onto an oligo- or polymeric backbone, we have proposed the notion of supramolecular glycomaterials in which a dense array of dye-functionalized glycosides are self-assembled onto a material through π–π stacking and/or electrostatic interaction, achieving the targeted imaging and eradication of cancer cells.14a,b Here, we developed a supramolecular 2D “glycomaterial” based on the self-assembly of HSA-based glycoproteins with 2D molybdenum disulphide (MoS2) for the asialoglycoprotein receptor (ASGPr)-targeted delivery of a theranostic cargo to liver cancer cells.
In recent years, a wide variety of 2D materials, owing to their large specific surface area and unique optical properties, have been developed as carriers and as photoactive agents for targeted delivery of therapeutic agents, leading to enhanced therapeutic outcomes for many diseases.15,16 Among the advanced materials developed, 2D MoS2 has been shown to be biocompatible with minimal toxicity to a number of healthy cells.17a However, the development of carbohydrate-modified MoS2 materials for targeted delivery of anticancer agents has been elusive. In this study, we used the “host–guest” inclusion of a fluorescent glycoprobe (Gal-DCM) and an anticancer drug (Maytansine) into the hydrophobic pockets of human serum albumin (HSA) to produce a multicomponent “glycoprotein”, which is then self-assembled with 2D MoS2. We show that the 2D glycomaterial is capable of targeted delivery of the theranostic cargo to Hep-G2 cells that highly expresses ASGPr on the cell surface (Scheme 1).
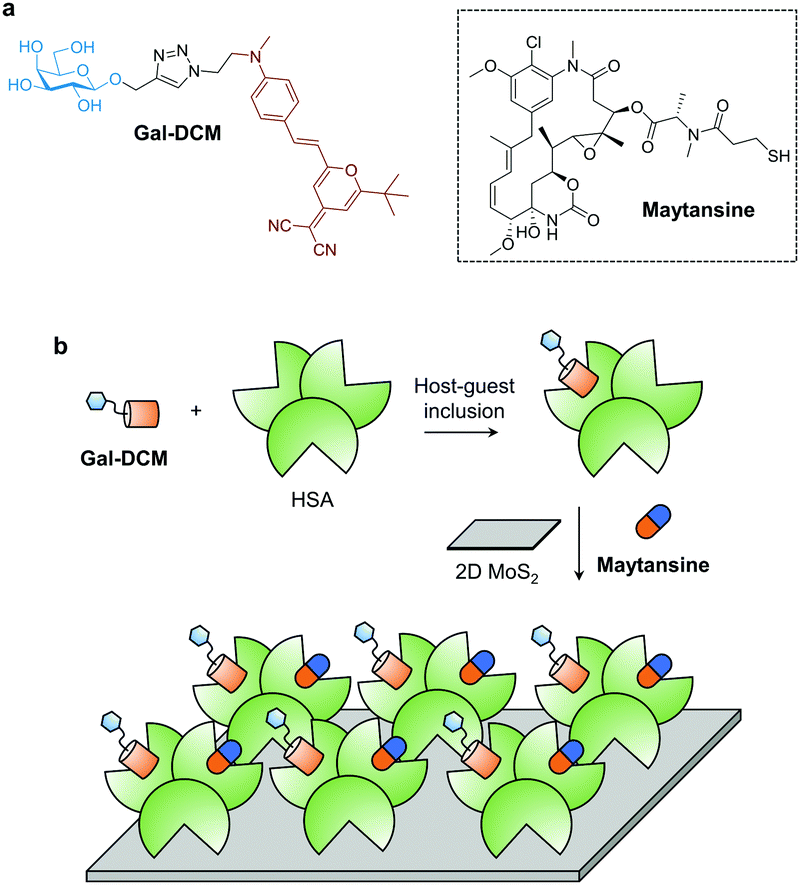 | ||
| Scheme 1 (a) Structure of galactosyl dicyanomethylene-4H-pyran (Gal-DCM) that targets the asialoglycoprotein receptor (ASGPr) of liver cancer cells, and Maytansine. (b) Construction of the supramolecular 2D glycomaterial by host–guest inclusion of Gal-DCM into human serum albumin (HSA), followed by the co-assembly of the resulting ensemble and Maytansine onto 2D molybdenum disulfide (2D MoS2) for targeted delivery of the drug to liver cancer cells (Hep-G2) that highly expresses ASGPr. | ||
Galactose (Gal) that serves as the targeting agent for ASPGr was coupled to a red-emitting dicyanomethylene-4H-pyran (DCM) dye using the click reaction, producing the glycoprobe Gal-DCM.14c In its typical UV-vis absorption and fluorescence emission spectra, the maximum absorbance and fluorescence emission peaks of Gal-DCM at ca. 460 and 630 nm, respectively, were observed (Fig. S1, ESI†). The glycoprobe was then bound to HSA through host–guest inclusion of DCM to the site 1 (IIA domain) of albumin.18 Subsequently, the resulting glycoprotein (Gal-DCM/HSA) was used to self-assemble with 2D MoS2 synthesized using a known exfoliation method14c in phosphate buffered saline (PBS, 0.01 M, pH 7.4) to form the 2D glycomaterial (Gal-DCM/HSA/2D MoS2). To study the morphology of the 2D glycomaterial, we used high-resolution transmission electron microscopy (HRTEM) and scanning electron microscopy (SEM) (Fig. S2, ESI†). 2D MoS2 alone was observed as thin-layered nanosheets. After the formation of the 2D glycomaterial, entangled fibril architectures corresponding to HSA were observed on the nanosheets. The size distribution of 2D MoS2, the glycoprotein and the 2D glycomaterial was measured using dynamic light scattering (DLS) (Fig. S3, ESI†). The diameter of the glycoprotein and 2D MoS2 ranged from 10–70 nm and 110–1000 nm, respectively. After self-assembly, the size increased to 150–2200 nm.
To elucidate the driving force by which Gal-DCM/HSA self-assembles with 2D MoS2, we used Raman spectroscopy and zeta potentiometry to characterize the scattering signals and potential of the materials generated, respectively. Typical Raman peaks centred at 378 cm−1 and 401 cm−1, which are assigned to the in-plane vibration (E12g) of Mo and S atoms and the reverse interlaminar vibration of two S atoms (A1g), respectively, were observed for both 2D MoS2 and the supramolecular glycomaterial (Fig. 1a). The ratio of E12g to A1g as used to characterize the changes in vibration level and thickness of the 2D MoS2 sheets. In the presence of the glycoprotein, the E12g/A1g ratio of the 2D MoS2 increased from 0.45 to 0.52, suggesting an increased degree of the in-plane Mo–S vibration and a decreased interlaminar vibration of the S atoms.19 This suggests that the coating of the glycoproteins onto the surface of 2D MoS2 increased the conjugation degree and thickness of the system. We also observed that the assembly of glycoproteins with 2D MoS2 increased the zeta potential from −37 mV to −25 mV (Fig. 1b), suggesting an electrostatic attraction between the two species. In addition, zeta potential measurement of the 2D glycomaterial at different pH showed that the material was relatively stable from pH 5.0 to 10.0, and especially at a physiological pH range of 6.0 to 8.0 (Fig. S4, ESI†).
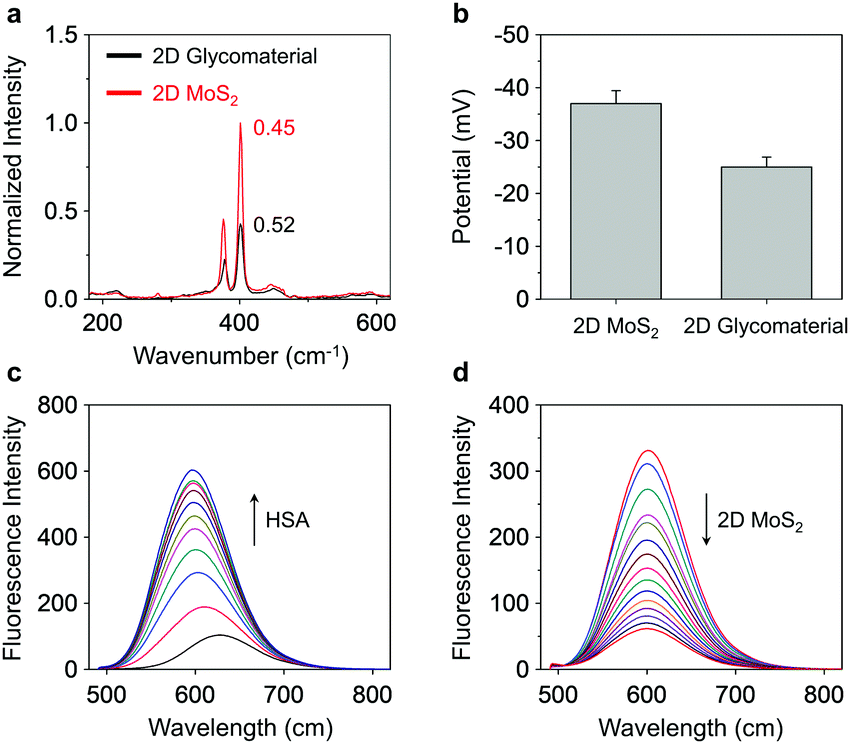 | ||
| Fig. 1 (a) Stacked Raman spectra of 2D MoS2 (1 μg mL−1) and 2D glycomaterial (Gal-DCM/Maytansine/HSA/2D MoS2 = 1 μM/20 nM/1 μM/1 μg mL−1). (b) Zeta potential of 2D MoS2 (1 μg mL−1) and 2D glycomaterial (Gal-DCM/Maytansine/HSA/2D MoS2 = 1 μM/20 nM/1 μM/1 μg mL−1). (c) Fluorescence enhancement of Gal-DCM (1 μM) with increasing HSA (0–50 μM; interval: 5 μM); excitation wavelength: 460 nm. (d) Fluorescence quenching of Gal-DCM/Maytansine/HSA (1 μM/20 nM/15 μM) with increasing 2D MoS2 (from the top curve to bottom: 0–28 μg mL−1; interval: 2 μg mL−1); excitation wavelength: 460 nm. | ||
Fluorescence spectroscopy was then used to characterize the supramolecular assemblies. We first determined that the addition of increasing concentrations of HSA to the Gal-DCM solution gradually enhanced the red emission (λmax = 600 nm) of DCM (Fig. 1c and Fig. S5a, ESI†). This suggests the inclusion of the dye to the hydrophobic cavity of the albumin, results in a significantly enhanced fluorescence as the hydrophobicity of the cavity of HSA supresses the intramolecular charge transfer (ICT) process of DCM.18 A competition assay was subsequently performed and indicated that pretreatment of a known site 1-binding agent (warfarin sodium)20 with HSA decreased the fluorescence of Gal-DCM in a concentration-dependent manner (Fig S5b, ESI†). In contrast, the pretreatment of dansyl-L-norvaline (a known site 2 (IIIA domain)-binding agent) with HSA did not cause the fluorescence of Gal-DCM to decrease (Fig S5c, ESI†), corroborating that the glycoprobe is predominantly included in the site 1 pocket of HSA. Using the same assay, we also determined that the preincubation of Maytansine with HSA did not result in a decrease in the fluorescence of Gal-DCM, suggesting that the binding domain of the fluorescent probe and that of the drug in HSA are not competitive (Fig. S5d, ESI†). Using isothermal titration calorimetry (ITC),21 the association constants (Ka) for Gal-DCM and Maytansine with HSA were determined to be approximately 2.1 × 105 and 1.5 × 105 L mol−1, respectively (Fig. S6, ESI†).
Next, a fluorescence titration was carried out for the fluorescent glycoprotein with 2D MoS2. We observed a gradually quenching of the fluorescence of DCM with increasing concentrations of 2D MoS2 (Fig. 1d). Which agrees with the quenching ability of 2D MoS2 for adjacent fluorescent dyes through energy and electron transfer mechanisms.17b The drug-loading capacity of the materials was determined by UV-vis spectroscopy (Fig. S7, ESI†). We observed a 20.8-fold greater Maytansine-loading of the supramolecular glycoprotein Gal-DCM/HSA (218 nM) than that of just 2D MoS2 (10 nM). Interestingly, the assembly of the glycoprotein to 2D MoS2 improved the Maytansine-loading amount further to 308 nM, which suggests that the unique architecture of the 2D glycomaterial offers more space for accommodating drug molecules.
Next, we evaluated the targeted cellular imaging ability of the 2D glycomaterial for Hep-G2 (human liver cancer cell that overexpresses ASGPr).14d A human triple-negative breast cancer cell line (MDA-MB-231) with minimal expression level of ASGPr was used as control. We determined that the incubation of 2D glycomaterial with the cells only led to the fluorescence production in Hep-G2 but not MDA-MB-231 cells. The receptor-dependent imaging effect of the 2D glycomaterial was evaluated via a competition assay. The pre-incubation of free D-galactose with Hep-G2 reduced the fluorescence of 2D glycomaterial in a concentration-dependent manner (Fig. 2). This suggests that selective imaging of the 2D glycomaterial is dependent on the galactose-ASGPr recognition. In addition, a decreased intracellular fluorescence from the 2D glycomaterial in Hep-G2 cells was observed, while no significant changes in MDA-MB-231 cells when the temperature was decreased from 37 °C to 4 °C (Fig. S8, ESI†), suggesting the cell uptake of the 2D glycomaterial is kinetically controlled via a receptor-mediated endocytic process.
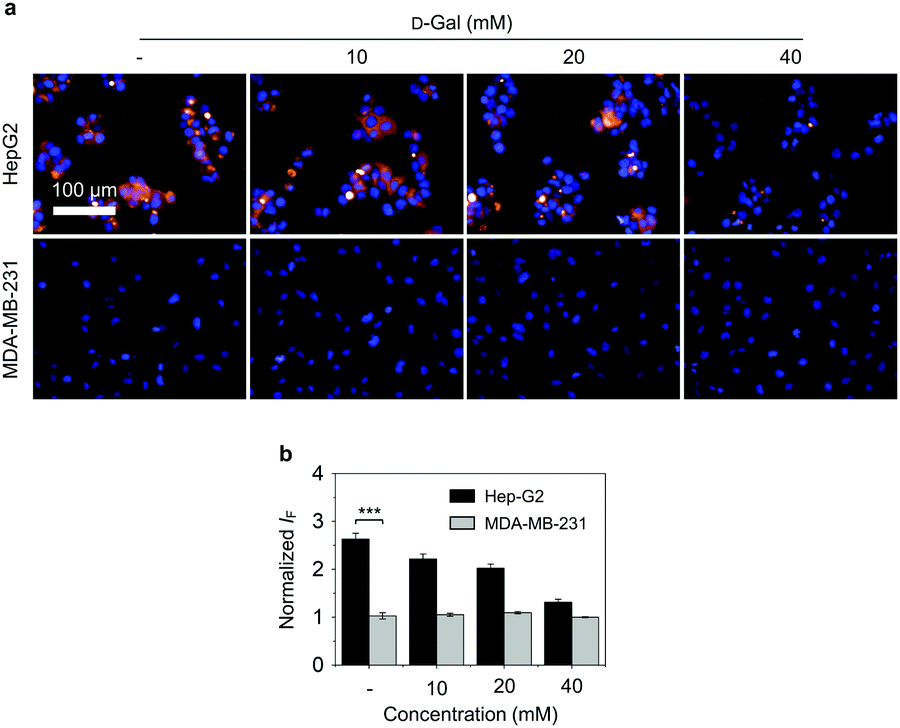 | ||
| Fig. 2 (a) Fluorescence imaging and (b) quantification of Hep-G2 (human liver cancer) and MDA-MB-231 (human breast cancer) cells after incubation with the 2D glycomaterial (Gal-DCM/HSA/2D MoS2 = 5 μM/5 μM/5 μg mL−1) without and with pretreatment of increasing free D-galactose (D-Gal) at the indicated concentrations. ***P < 0.001. S.D. means standard deviation (n = 3). The excitation and emission channel used is 460–490 nm and 580–650 nm, respectively. | ||
Subsequently, the potential of the 2D glycomaterial as a drug delivery carrier was evaluated. It has been reported that Maytansine could be included into site 1 of HSA resulting in enhanced biosafety of Maytansine-based therapy in vivo.11b A preliminary cell viability assay indicated a ca. 56%, 53% and 35% reduced IC50 (half maximal inhibitory concentration) of the 2D glycomaterial-loaded Maytansine (IC50 = 0.15 μM) compared to Maytansine (IC50 = 0.34 μM), Maytansine/HSA (IC50 = 0.32 μM) and Gal-DCM/Maytansine/HSA (IC50 = 0.23 μM), respectively (Fig. S9, ESI†), suggesting the presence of the galactosyl targeting agent significantly enhanced the ASGPr-mediated endocytosis of the therapeutic system.
Next, a double-staining assay was carried out to corroborate the targeted cytotoxicity of the delivery system. Hoechst 33342 and EasyProbe Green 488 were used to stain the total and dead cells, respectively. As shown in Fig. S10 (ESI†), when cells were treated with Maytansine-loaded 2D glycomaterial, a strong fluorescence emission assigned to EasyProbe Green 488 was observed in Hep-G2 but not in MDA-MB-231 cells. This corroborates the selective internalization of the galactose-modified 2D material for ASGPr-targeted delivery of cytotoxic drugs inducing cell death. Maytansine was reported to induce cell apoptosis through blocking the polymerisation of microtubule proteins, which arrests cells in the G2/M phase of the cell cycle, thereby inhibiting mitosis.11c Thus, the Annexin V-mCherry Apoptosis Detection Kit was used to test whether the Maytansine-loaded 2D glycomaterial kills cancer cells via an apoptotic pathway. The result indicated that the incubation of Maytansine alone with Hep-G2 cells caused a slight increase in fluorescence, as did the assembly of the drug with HSA. A further increase in fluorescence was seen when Maytansine was co-assembled with HSA and 2D MoS2. The incorporation of Gal-DCM into the system further increased the Annexin V-mCherry fluorescence, which suggests the receptor-targeting delivery of Maytansine resulted in cell apoptosis (Fig. S11, ESI†).
To summarize, we have constructed a simple 2D glycomaterial for targeted delivery of Maytansine to Hep-G2 cells with enhanced expression of ASGPr, leading to cell apoptosis. This study offers insight into the simple construction of sugar-based supramolecular materials for the targeted delivery of commercial cytotoxic drugs to cancer cells.22
X.-P. H. thanks the National Natural Science Foundation of China (No. 21788102, 91853201), the Shanghai Science and Technology Committee (No. 19410712600), the Shanghai Municipal Science and Technology Major Project (No. 2018SHZDZX03), the Fundamental Research Funds for the Central Universities (222201717003) and the Programme of Introducing Talents of Discipline to Universities (B16017) for financial support. H.-H. H. thanks the National Natural Science Foundation of China (No. 22107029) and Project funded by China Postdoctoral Science Foundation (No. 2020M681196). T. D. J. wishes to thank the Royal Society for a Wolfson Research Merit Award and the Open Research Fund of the School of Chemistry and Chemical Engineering, Henan Normal University for support (2020ZD01).
Conflicts of interest
There are no conflicts to declare.Notes and references
- J.-G. Chen, J. Zhu, Y.-H. Zhang, Y.-S. Chen, J.-H. Lu, Y.-R. Zhu, H.-Z. Chen, A.-G. Shen, G.-R. Wang, J. D. Groopman and T. W. Kensler, PeerJ, 2021, 9, e10600–e10600 CrossRef PubMed.
- M. Le Grazie, M. R. Biagini, M. Tarocchi, S. Polvani and A. Galli, World J. Hepatol., 2017, 9, 907–920 CrossRef PubMed.
- F. Zhou, W. Shang, X. Yu and J. Tian, Med. Res. Rev., 2018, 38, 741–767 CrossRef CAS PubMed.
- H.-H. Han, Y.-J. Qiu, Y.-Y. Shi, W. Wen, X.-P. He, L.-W. Dong, Y.-X. Tan, Y.-T. Long, H. Tian and H.-Y. Wang, Theranostics, 2018, 8, 3268–3274 CrossRef CAS PubMed.
- Y. Fu, D. J. Urban, R. R. Nani, Y. F. Zhang, N. Li, H. Fu, H. Shah, A. P. Gorka, R. Guha, L. Chen, M. D. Hall, M. J. Schnermann and M. Ho, Hepatology, 2019, 70, 563–576 CrossRef CAS PubMed.
- Y. C. Lee and R. T. Lee, Acc. Chem. Res., 1995, 28, 321–327 CrossRef CAS.
- Q.-L. He, I. Minn, Q. Wang, P. Xu, S. A. Head, E. Datan, B. Yu, M. G. Pomper and J. O. Liu, Angew. Chem., Int. Ed., 2016, 55, 12035–12039 CrossRef CAS PubMed.
- M. Lu, M. Zhang, B. Hu and Y. Huang, in Design and Delivery of SiRNA Therapeutics, ed. H. J. Ditzel, M. Tuttolomondo and S. Kauppinen, Springer US, New York, NY, 2021, pp. 77–100 DOI:10.1007/978-1-0716-1298-9_6.
- (a) H.-H. Han, A. C. Sedgwick, Y. Shang, N. Li, T. Liu, B.-H. Li, K. Yu, Y. Zang, J. T. Brewster, M. L. Odyniec, M. Weber, S. D. Bull, J. Li, J. L. Sessler, T. D. James, X.-P. He and H. Tian, Chem. Sci., 2020, 11, 1107–1113 RSC; (b) X.-L. Hu, Q. Cai, J. Gao, R. A. Field, G.-R. Chen, N. Jia, Y. Zang, J. Li and X.-P. He, ACS Appl. Mater. Interfaces, 2019, 11, 22181–22187 CrossRef CAS PubMed.
- (a) G. Yang, S. Z. F. Phua, W. Q. Lim, R. Zhang, L. Feng, G. Liu, H. Wu, A. K. Bindra, D. Jana, Z. Liu and Y. Zhao, Adv. Mater., 2019, 31, 1901513 CrossRef PubMed; (b) Z. Yang, Q. Chen, J. Chen, Z. Dong, R. Zhang, J. Liu and Z. Liu, Small, 2018, 14, 1803262 CrossRef.
- (a) Y. R. Zheng, K. Suntharalingam, T. C. Johnstone, H. Yoo, W. Lin, J. G. Brooks and S. J. Lippard, J. Am. Chem. Soc., 2014, 136, 8790–8798 CrossRef CAS PubMed; (b) H. Wang, J. Wu, L. Xu, K. Xie, C. Chen and Y. Dong, Chem. Commun., 2017, 53, 2618–2621 RSC; (c) S. Remillard, L. I. Rebhun, G. A. Howie and S. M. Kupchan, Science, 1975, 189, 1002–1005 CrossRef CAS PubMed.
- C. R. Bertozzi and L. L. Kiessling, Science, 2001, 291, 2357–2364 CrossRef CAS PubMed.
- J. J. Lundquist and E. J. Toone, Chem. Rev., 2002, 102, 555–578 CrossRef CAS PubMed.
- (a) H.-L. Zhang, X.-L. Wei, Y. Zang, J.-Y. Cao, S. Liu, X.-P. He, Q. Chen, Y.-T. Long, J. Li, G.-R. Chen and K. Chen, Adv. Mater., 2013, 25, 4097–4101 CrossRef CAS PubMed; (b) H.-H. Han, C.-Z. Wang, Y. Zang, J. Li, T. D. James and X.-P. He, Chem. Commun., 2017, 53, 9793–9796 RSC; (c) D.-K. Ji, Y. Zhang, Y. Zang, J. Li, G.-R. Chen, X.-P. He and H. Tian, Adv. Mater., 2016, 28, 9356–9363 CrossRef CAS PubMed; (d) J. Zhang, Y. Fu, H.-H. Han, Y. Zang, J. Li, X.-P. He, B. L. Feringa and H. Tian, Nat. Commun., 2017, 8, 987 CrossRef PubMed.
- X. Wang, X. Zhong, J. Li, Z. Liu and L. Cheng, Chem. Soc. Rev., 2021, 50, 8669–8742 RSC.
- W.-C. Geng, J. L. Sessler and D.-S. Guo, Chem. Soc. Rev., 2020, 49, 2303–2315 RSC.
- (a) X. P. He and H. Tian, Small, 2016, 12, 144–160 CrossRef CAS PubMed; (b) Y. Chen, C. Tan, H. Zhang and L. Wang, Chem. Soc. Rev., 2015, 44, 2681–2701 RSC.
- J. Fan, W. Sun, Z. Wang, X. Peng, Y. Li and J. Cao, Chem. Commun., 2014, 50, 9573–9576 RSC.
- P. T. K. Loan, W. Zhang, C.-T. Lin, K.-H. Wei, L.-J. Li and C.-H. Chen, Adv. Mater., 2014, 26, 4838–4844 CrossRef CAS PubMed.
- I. Petitpas, A. A. Bhattacharya, S. Twine, M. East and S. Curry, J. Biol. Chem., 2001, 276, 22804–22809 CrossRef CAS PubMed.
- (a) W. R. Archer and M. D. Schulz, Soft Matter, 2020, 16, 8760–8774 RSC; (b) J. M. R. Duff, J. Grubbs and E. E. Howell, JoVE, 2011, e2796, DOI:10.3791/2796.
- W.-T. Dou, H.-H. Han, A. C. Sedgwick, G.-B. Zhu, Y. Zang, X.-R. Yang, J. Yoon, T. D. James, J. Li and X.-P. He, Sci. Bull., 2022 DOI:10.1016/j.scib.2022.01.014.
Footnotes |
| † Electronic supplementary information (ESI) available: Additional figures, experimental details. See DOI: 10.1039/d1cc06809a |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2022 |