
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHydride reduction of o-(fluorosilyl)benzodifluorides for subsequent C–F transformations†‡
Rika
Idogawa
a,
Akihiro
Kobayashi
ab,
Youngchan
Kim
a,
Ken
Shimomori
a,
Takamitsu
Hosoya
 a and
Suguru
Yoshida
*b
a and
Suguru
Yoshida
*b
aLaboratory of Chemical Bioscience, Institute of Biomaterials and Bioengineering, Tokyo Medical and Dental University (TMDU), 2-3-10 Kanda-Surugadai, Chiyoda-ku, Tokyo 101-0062, Japan
bDepartment of Biological Science and Technology, Faculty of Advanced Engineering, Tokyo University of Science, 6-3-1 Niijuku, Katsushika-ku, Tokyo 125-8585, Japan. E-mail: s-yoshida@rs.tus.ac.jp
First published on 15th February 2022
Abstract
An efficient method for sequential C–F transformations of o-hydrosilyl-substituted benzotrifluorides is disclosed. A key to the success is hydride reduction of o-fluorosilyl-substituted difluoromethylenes prepared by a single C–F transformation of o-hydrosilyl-substituted benzotrifluorides. We succeeded in further C–F transformations via hydride abstraction of the resulting o-hydrosilyl group, enabling us to synthesize a wide variety of organofluorine compounds.
Organofluorine compounds are of great importance in broad research fields including pharmaceutical sciences, agrochemistry, and materials chemistry.1 A wide variety of fluorinating reagents have been developed so far for synthesizing a wide range of organofluorines from halides and alcohols (Fig. 1A).2 Despite the remarkable improvements in organofluorine chemistry enhancing the availability of organofluorines, it is not easy to synthesize highly functionalized benzyl fluoride derivatives due to the limited fluorination reactions.3
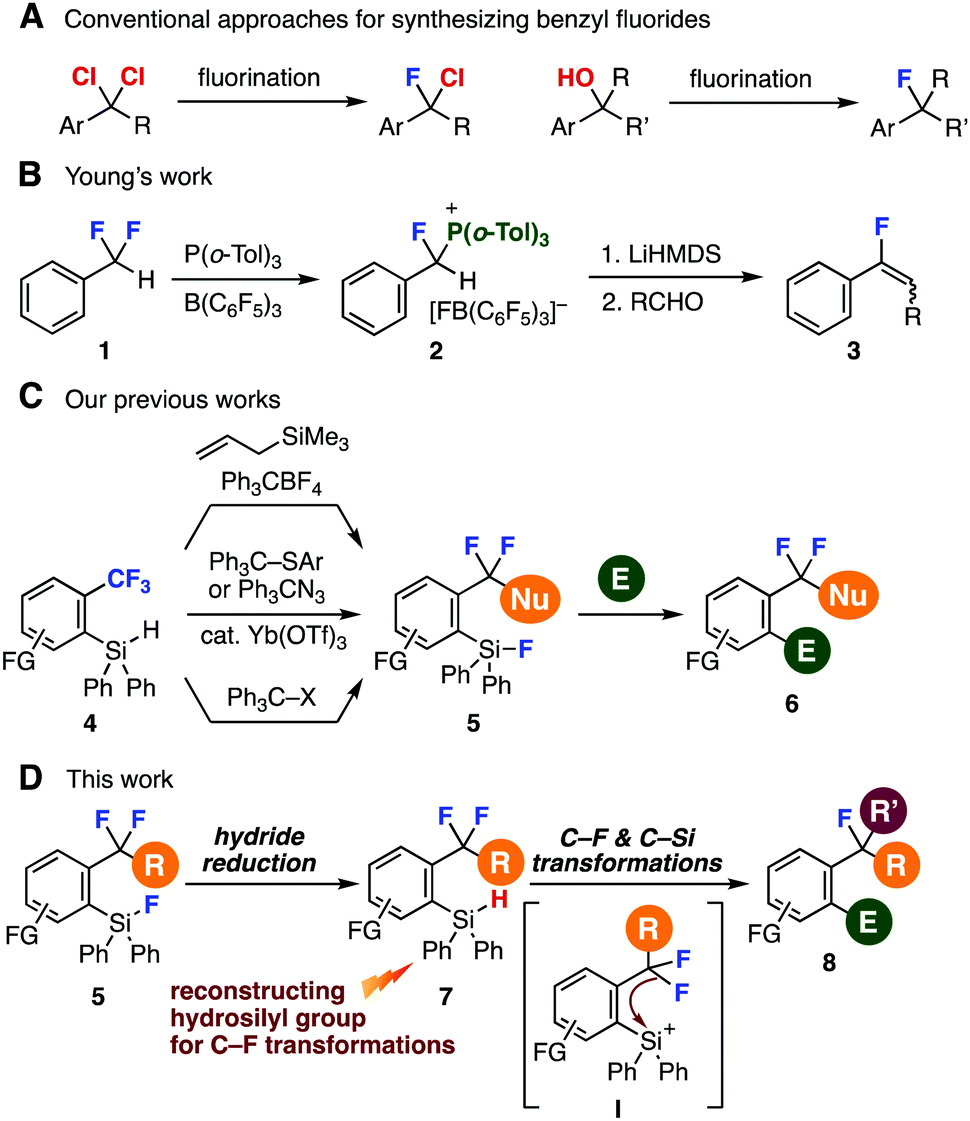 | ||
| Fig. 1 Backgrounds and an abstract of this study. (A) Conventional methods to synthesize benzyl fluorides. (B) Young's work. (C) Our previous studies. (D) This work. | ||
Modern studies of selective C–F transformations significantly expanded the accessibility of organofluorines (Fig. 1B and C).4,5 For example, in 2018, Young and coworkers succeeded in a single C–F transformation of α,α-difluorotoluene (1) with a frustrated Lewis pair between tri(o-tolyl)phosphine and tris(pentafluorophenyl)borane realizing facile synthesis of phosphonium salt 2, which served in the preparation of fluoroalkenes by the Wittig reaction with aldehydes (Fig. 1B).6 Our recent achievements from 2016 on single C–F transformations of benzotrifluorides 4 enabled to synthesize difluoromethylenes 5 through hydride abstraction of o-hydrosilyl group (Fig. 1C).7 Herein, we disclose a new method to synthesize highly functionalized benzyl fluorides 8 from o-fluorosilyl-substituted difluoromethylenes 5 by C–F and C–Si transformations (Fig. 1D). A key to the success was efficient reconstruction of hydrosilyl group from fluorosilyl groups, which allowed further C–F transformations via silyl cation intermediate I generated by hydride abstraction.
First, we examined hydride reduction of o-fluorosilyl-substituted difluoromethylene 5a using various metal hydrides (Table 1).8 As a result, o-hydrosilyl-substituted benzodifluoride 7a was synthesized quantitatively with lithium aluminum hydride (LAH) without damaging labile difluoromethylene moiety8 and C–Si cleavage (entry 1). In sharp contrast, hydride reduction of fluorosilane 5a resulted in failure when using sodium borohydride, diisobutylaluminum hydride, lithium triisobutylborohydride, or lithium triethylborohydride due to undesired decomposition (entries 2–5). The efficient LAH reduction of fluorosilane 7a took place in 1.0 mmol scale, clearly showing the good scalability.
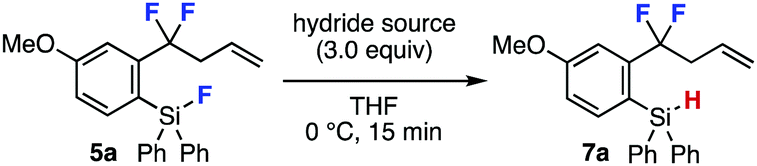
A wide range of o-fluorosilyl-substituted benzodifluorides 5 successfully participated in the hydride reduction to afford o-(hydrosilyl)benzodifluorides 7 (Fig. 2). For example, hydrosilanes 7b and 7c having electron-rich thienyl and electron-deficient 4-(trifluoromethyl)phenyl group, respectively, were efficiently prepared by LAH reduction. Hydride reduction furnishing bromo-substituted silane 7d also proceeded smoothly leaving the bromo group untouched. The reduction of fluorodimethylsilyl group instead of fluorodiphenylsilyl group proceeded and required longer reaction time affording hydrosilane 7e in good yield,9 where the dimethylsilyl group can serve in the C–F transformations.7a Furthermore, efficient synthesis of 7f bearing an acidic allylic proton was achieved without damaging difluoromethylene moiety. We succeeded in the preparation of difluorobenzyl sulfide 7g and chlorides 7h and 7i in good yields. Also, difluorobenzyl bromide 7j was synthesized in moderate yield. Unfortunately, the synthesis of difluorobenzyl p-toluenesulfonate 7k resulted in failure due to the labile sulfonate ester moiety.
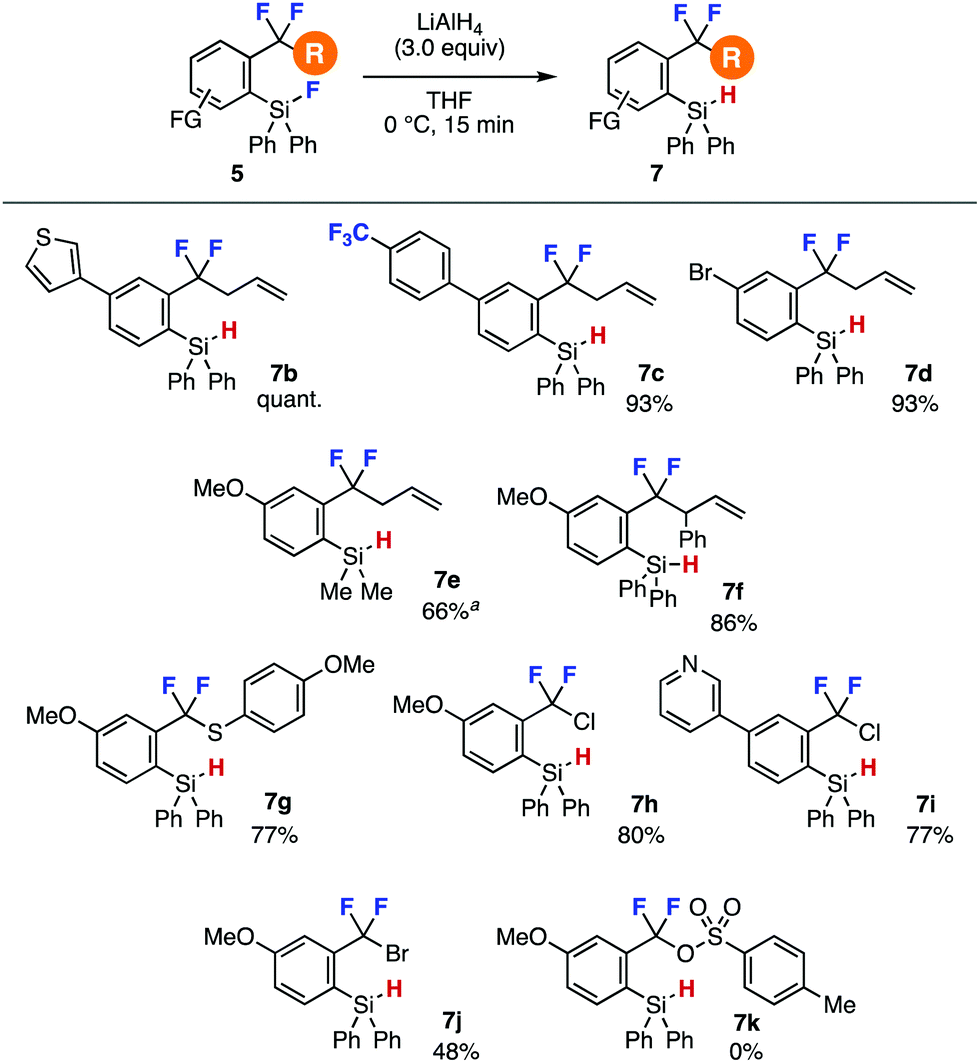 | ||
| Fig. 2 Syntheses of o-(hydrosilyl)benzodifluorides 7. See the ESI‡ for details. aThe reaction time was 2 h. | ||
The reconstructed hydrosilyl group of difluoromethylenes served in further C–F transformations (Fig. 3). Indeed, single C–F chlorination of difluoromethylene 7a with trityl chloride in chlorobenzene and 1,1,1,3,3,3-hexafluoro-2-propanol (HFIP)10 through benzyl cation intermediate II proceeded efficiently to afford highly functionalized benzyl fluoride 8a in good yield (Fig. 3A).7c Various benzyl fluorides 8b–8e were synthesized through single C–F transformations of difluoromethylenes 7a–7d having a range of functional groups (Fig. 3B). For instance, C–F chlorination products 8b–8d were prepared from 7b–7d with trityl chloride in moderate to good yields without damaging a wide variety of functionalities such as fluoro, chloro, allyl, fluorosilyl, trifluoromethyl, thienyl, and bromo groups. It is worth noting that benzyl chloride 8c was prepared selectively without C–F chlorination of trifluoromethyl group. Further C–F allylation of benzodifluoride 7a also took place to afford 8e in moderate yield.7a When we attempted Yb-catalyzed C–F thiolation of benzodifluoride 7a with 4-tolyl trityl sulfide,7b benzyl fluoride 8f was not obtained, showing the different reactivity of fluorobenzyl cation II generated from 7a to difluorobenzyl cation intermediates in our previous reports.7 Owing to the great importance of organofluorine chemistry, successes in the synthesis of highly functionalized benzyl fluorides 8a–8e obviously indicated the significant potential of sequential transformations via reconstruction of the hydrosilyl group.
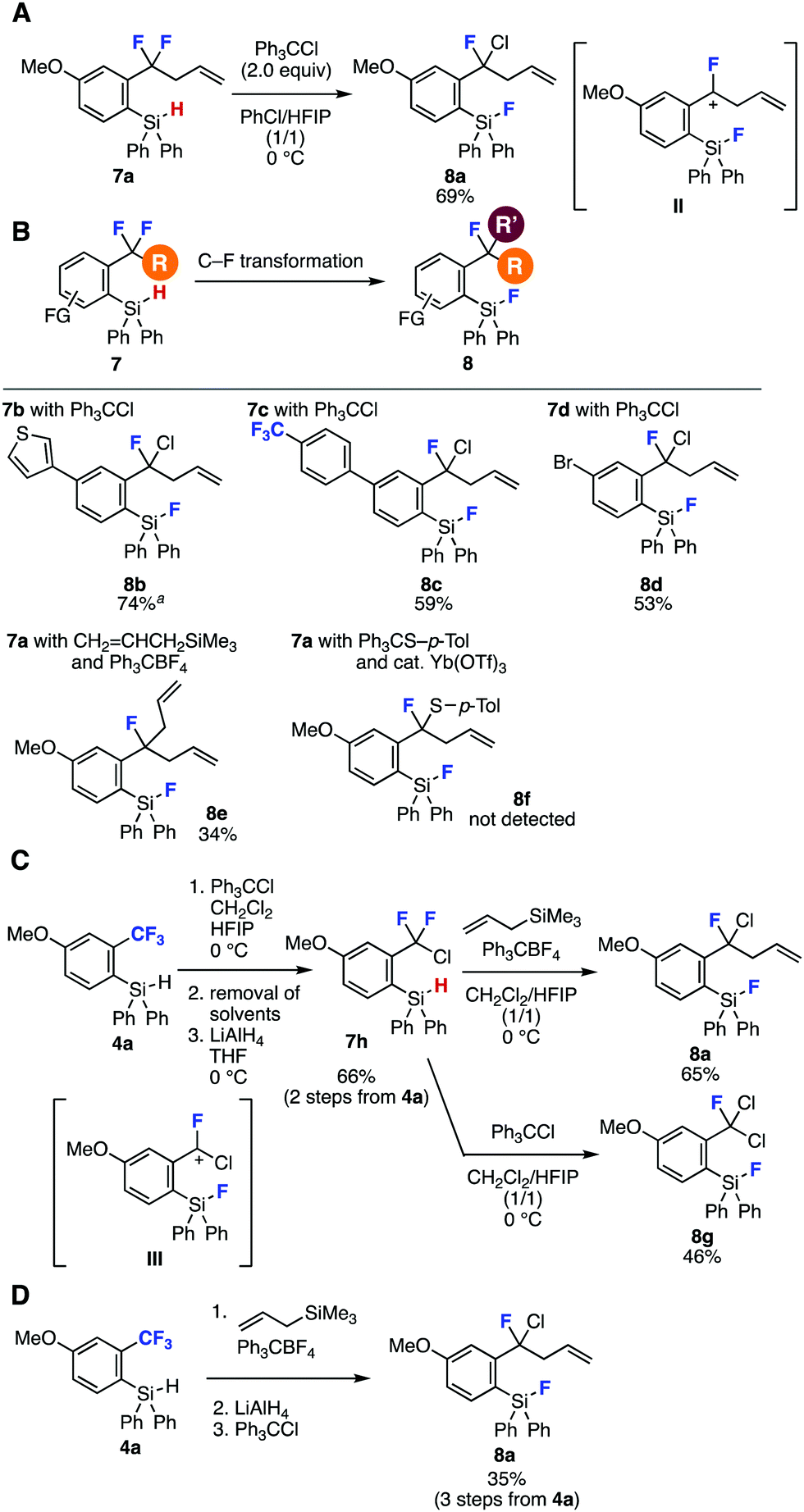 | ||
| Fig. 3 Transformations of o-(hydrosilyl)benzodifluorides 7. See the ESI‡ for details. Isolated yields are shown otherwise noted. aNMR yield. (A) C–F chlorination of 7a. (B) Scope of benzyl fluoride synthesis. (C) Sequential C–F transformations of 4avia difluorobenzyl chloride 7h. (D) C–F transformations of 4a with a single silica-gel column chromatography purification. | ||
Facile synthesis of α,α-difluorobenzyl chloride 7h by a single C–F chlorination and subsequent LAH reduction was achieved from benzotrifluoride 4a in a one-pot manner via removal of solvents under reduced pressure (Fig. 3C). Benzyl fluoride 8a was prepared in good yield also from α,α-difluorobenzyl chloride 7h by a single C–F allylation provably via α-chloro-α-fluorobenzyl cation III. Second C–F chlorination took place smoothly to provide α,α-dichlorobenzyl fluoride 8g in moderate yield. Moreover, we succeeded in the preparation of benzyl fluoride 8a from benzotrifluoride 4a in a three-step, single purification procedure (Fig. 3D). Indeed, C–F allylation of 4a followed by LAH reduction using the crude product and subsequent C–F chlorination provided benzyl fluoride 8a in moderate yield.
Succeeding transformations of highly functionalized benzyl fluoride 8a realized facile synthesis of organofluorines involving 1-aryl-1-fluoro-1,3-butadiene 10a (Fig. 4A). For example, silver mediated C–Si bromination11 of fluorosilane 8a furnished bromide 9a in moderate yields leaving various reactive functional groups intact (Fig. 4A, upper). Treatment of 9a with cesium carbonate in dimethyl sulfoxide (DMSO) at 120 °C provided 1,3-butadiene 10a in high yield with good Z selectivity.12 Although 1,3-butadiene 10a was found to be labile under various conditions such as acidic or basic aqueous conditions, it is worthy to note that 1,3-butadiene 10a was synthesized by heating 9a in DMSO in the presences of cesium carbonate in high yield.13 In contrast, no diene formation was observed when boiling benzyl fluoride 9a in the presence of cesium carbonate in toluene. When we treated o-fluorosilyl-substituted benzyl fluoride 8a with 4-iodotoluene in the presence of silver oxide and a catalytic amount of palladium catalyst, biaryl 11 was obtained in good yield via the Hiyama cross-coupling and dehydrochlorination (Fig. 4A, lower).14
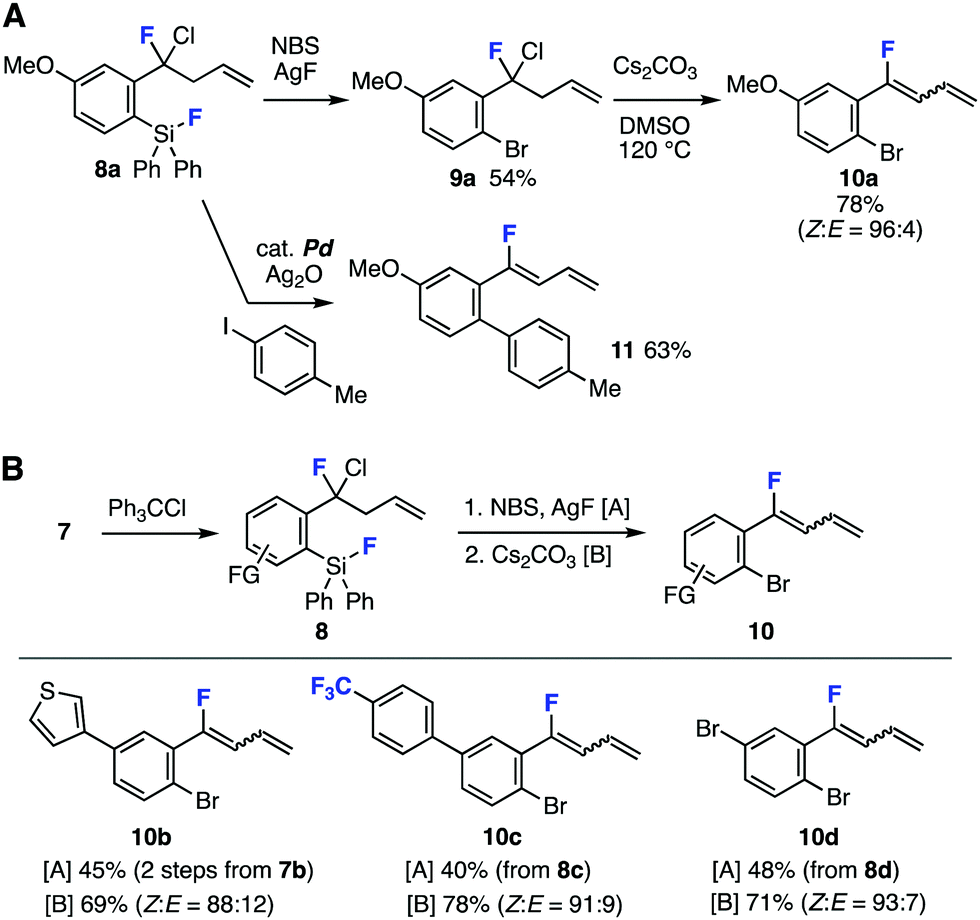 | ||
| Fig. 4 Transformations of benzyl fluorides 8. (A) Synthesis of various organofluorines from 8a. (B) Fluorobutadiene synthesis. | ||
A range of 1-aryl-1-fluoro-1,3-butadiens 10b–10d were successfully prepared from 8b–8d by C–Si bromination and following 1,3-butadiene formation (Fig. 4B). Desilylbromination of benzyl fluoride 8b prepared by C–F chlorination of difluoromethylene 7b and following dehydrochlorination furnished thienyl-substituted 1-fluoro-1,3-butadiene 10b. Also, fluorobutadienes 10c and 10d were successfully synthesized from difluoromethylenes 7c and 7d having electron-deficient aromatic ring and transformable bromo group, respectively, in good yields. Since 1,3-butadienes are versatile building blocks in synthetic organic chemistry,15 this unique method to prepare functionalized fluorobutadienes will serve to synthesize a broad range of organofluorines.
In summary, we accomplished the synthesis of a wide variety of organofluorines through LAH reduction of o-(fluorosilyl)benzodifluorides. A broad range of benzyl fluorides and 1-aryl-1-fluoro-1,3-butadienes were successfully prepared by C–F transformations of o-(fluorosilyl)benzodifluorides and following transformations. Further studies such as diversifications of 1-aryl-1-fluoro-1,3-butadienes are ongoing in our laboratory.
The authors thank Dr Yuki Sakata at Tokyo Medical and Dental University for HRMS analyses. This work was supported by JSPS KAKENHI Grant Number JP19K05451 (C; S. Y.); the Naito Foundation (S. Y.); the Japan Agency for Medical Research and Development (AMED) under Grant Number JP21am0101098 (Platform Project for Supporting Drug Discovery and Life Science Research, BINDS); and the Cooperative Research Project of Research Center for Biomedical Engineering.
Conflicts of interest
There are no conflicts to declare.Notes and references
- (a) S. Purser, P. R. Moore, S. Swallow and V. Gouverneur, Chem. Soc. Rev., 2008, 37, 320 RSC; (b) E. P. Gillis, K. J. Eastman, M. D. Hill, D. J. Donnelly and N. A. Meanwell, J. Med. Chem., 2015, 58, 8315 CrossRef CAS PubMed.
- T. Hiyama and H. Yamamoto, Organofluorine Building Blocks, in Organofluorine Compounds, ed. H. Yamamoto, Springer, Berlin, 2000, pp, 77–118 Search PubMed.
- (a) K. Fuchibe, R. Oki, H. Hatta and J. Ichikawa, Chem. – Eur. J., 2018, 24, 17932 CrossRef CAS PubMed; (b) K. Komoda, R. Iwamoto, M. Kasumi and H. Amii, Molecules, 2018, 23, 3292 CrossRef PubMed; (c) S. Kawamura, C. J. Henderson, Y. Aoki, D. Sekine, S. Kobayashi and M. Sodeoka, Chem. Commun., 2018, 54, 11276 RSC; (d) X. Zeng, W. Yan, S. B. Zacate, T.-H. Chao, X. Sun, Z. Cao, K. G. E. Bradford, M. Paeth, S. B. Tyndall, K. Yang, T.-C. Kuo, M.-J. Cheng and W. Liu, J. Am. Chem. Soc., 2019, 141, 11398 CrossRef CAS PubMed; (e) N. Hisano, D. Kimura and K. Mori, Chem. Lett., 2019, 48, 771 CrossRef CAS; (f) C. F. Meyer, S. M. Hell, A. Misale, A. A. Trabanco and V. Gouverneur, Angew. Chem., Int. Ed., 2019, 58, 8829 CrossRef CAS PubMed; (g) L. Tang, Z.-Y. Liu, W. She and C. Feng, Chem. Sci., 2019, 10, 8701 RSC; (h) E. Miller, S. Kim, K. Gibson, J. S. Derrick and F. D. Toste, J. Am. Chem. Soc., 2020, 142, 8946 CrossRef CAS PubMed; (i) X. Jiang, D. Meyer, D. Baran, M. A. C. González and K. J. Szabó, J. Org. Chem., 2020, 85, 8311 CrossRef CAS PubMed; (j) L. Liao, R. An, H. Li, Y. Xu, J.-J. Wu and X. Zhao, Angew. Chem., Int. Ed., 2020, 59, 11010 CrossRef CAS PubMed; (k) Q. Xie, Z. Zhu, L. Li, C. Ni and J. Hu, Chem. Sci., 2020, 11, 276 RSC; (l) M. Cloutier, M. Mamone and J.-F. Paquin, Chem. Commun., 2020, 56, 5969 RSC; (m) H. Kadri, T. E. Taher, Q. Xu, M. Sharif, E. Ashby, R. T. Bryan, B. E. Willcox and Y. Mehellou, J. Med. Chem., 2020, 63, 11258 CrossRef CAS PubMed; (n) W. Wang, P. Wang, Q. Zhang, P. Du, J. Zhang, H. Deng and H. Jiang, Tetrahedron, 2020, 76, 131477 CrossRef CAS; (o) A. Tarui, M. Ueo, M. Morikawa, M. Tsuta, S. Iwasaki, N. Morishita, Y. Karuo, K. Sato, K. Kawai and M. Omote, Synthesis, 2020, 3657 CAS; (p) K. Kikushima, Y. Etou, R. Kamura, I. Takeda, H. Ito, M. Ohashi and S. Ogoshi, Org. Lett., 2020, 22, 8167 CrossRef CAS PubMed; (q) H. Fang, Q. He, G. Liu and Z. Huang, Org. Lett., 2020, 22, 9298 CrossRef CAS PubMed; (r) P. Wang, P. Du, Q. Sun, J. Zhang, H. Deng and H. Jiang, Org. Biomol. Chem., 2021, 19, 2023 RSC; (s) S.-Y. He, X.-W. Yan, H.-Y. Tu and X.-G. Zhang, Org. Chem. Front., 2021, 8, 4746 RSC.
- (a) F. Jaroschik, Chem. – Eur. J., 2018, 24, 14572 CrossRef CAS PubMed; (b) J.-D. Hamel and J.-F. Paquin, Chem. Commun., 2018, 54, 10224 RSC; (c) D. R. Carvalho and A. H. Christian, Org. Biomol. Chem., 2021, 19, 947 RSC; (d) G. Yan, K. Qiu and M. Guo, Org. Chem. Front., 2021, 8, 3915 RSC; (e) H.-J. Ai, X. Ma, Q. Song and X.-F. Wu, Sci. China: Chem., 2021, 64, 1630 CrossRef CAS.
- (a) H. Dang, A. M. Whittaker and G. Lalic, Chem. Sci., 2016, 7, 505 RSC; (b) I. Mallov, A. J. Ruddy, H. Zhu, S. Grimme and D. W. Stephan, Chem. – Eur. J., 2017, 23, 17692 CrossRef CAS PubMed; (c) S. B. Munoz, C. Ni, Z. Zhang, F. Wang, N. Shao, T. Mathew, G. A. Olah and G. K. S. Prakash, Eur. J. Org. Chem., 2017, 2322 CrossRef CAS; (d) K. Chen, N. Berg, R. Gschwind and B. König, J. Am. Chem. Soc., 2017, 139, 18444 CrossRef CAS PubMed; (e) H. Wang and N. T. Jui, J. Am. Chem. Soc., 2018, 140, 163 CrossRef CAS PubMed; (f) D. B. Vogt, C. P. Seath, H. Wang and N. T. Jui, J. Am. Chem. Soc., 2019, 141, 13203 CrossRef CAS PubMed; (g) C. Luo and J. S. Bandar, J. Am. Chem. Soc., 2019, 141, 14120 CrossRef CAS PubMed; (h) D. Mandal, R. Gupta, A. K. Jaiswal and R. D. Young, J. Am. Chem. Soc., 2020, 142, 2572 CrossRef CAS PubMed; (i) H. Iwamoto, H. Imiya, M. Ohashi and S. Ogoshi, J. Am. Chem. Soc., 2020, 142, 19360 CrossRef CAS PubMed; (j) M. Ikeda, T. Matsuzawa, T. Morita, T. Hosoya and S. Yoshida, Chem. – Eur. J., 2020, 26, 12333 CrossRef CAS PubMed; (k) R. Gupta, D. Mandal, A. K. Jaiswal and R. D. Young, Org. Lett., 2021, 23, 1915 CrossRef CAS PubMed; (l) N. Sugihara, K. Suzuki, Y. Nishimoto and M. Yasuda, J. Am. Chem. Soc., 2021, 143, 9308 CrossRef CAS PubMed; (m) K. I. Burton, I. Elser, A. E. Waked, T. Wagener, R. J. Andrews, F. Glorius and D. W. Stephan, Chem. – Eur. J., 2021, 27, 11730 CrossRef CAS PubMed; (n) Y.-C. Luo, F.-F. Tong, Y. Zhang, C.-Y. He and X. Zhang, J. Am. Chem. Soc., 2021, 143, 13971 CrossRef CAS PubMed; (o) S. Mkrtchyan, M. Jakubczyk, S. Lanka, M. Yar, K. Ayub, M. Shkoor, M. Pittelkow and V. O. Iaroshenko, Adv. Synth. Catal., 2021, 363, 5448 CrossRef CAS.
- D. Mandal, R. Gupta and R. D. Young, J. Am. Chem. Soc., 2018, 140, 10682 CrossRef CAS PubMed.
- (a) S. Yoshida, K. Shimomori, Y. Kim and T. Hosoya, Angew. Chem., Int. Ed., 2016, 55, 10406 CrossRef CAS PubMed; (b) Y. Kim, K. Kanemoto, K. Shimomori, T. Hosoya and S. Yoshida, Chem. – Eur. J., 2020, 26, 6136 CrossRef CAS PubMed; (c) R. Idogawa, Y. Kim, K. Shimomori, T. Hosoya and S. Yoshida, Org. Lett., 2020, 22, 9292 CrossRef CAS PubMed.
- (a) V. H. T. Chang and J. Y. Corey, J. Organomet. Chem., 1980, 190, 217 CrossRef CAS; (b) C. Eaborn and D. E. Reed, J. Chem. Soc., Perkin Trans. 2, 1985, 1687 RSC.
- When the reaction was performed for 15 min, hydrosilane 7e was obtained in 24% yield along with 76% recovery of fluorosilane 5e.
- (a) J. Ichikawa, S. Miyazaki, M. Fujiwara and T. Minami, J. Org. Chem., 1995, 60, 2320 CrossRef CAS; (b) J.-P. Bégué, D. Bonnet-Delpon and B. Crousse, Synlett, 2004, 18 Search PubMed; (c) T. Dohi, N. Yamaoka and Y. Kita, Tetrahedron, 2010, 66, 5775 CrossRef CAS.
- B. Su and J. F. Hartwig, J. Am. Chem. Soc., 2017, 139, 12137 CrossRef CAS PubMed.
- T. J. O’Connor and F. D. Toste, ACS Catal., 2018, 8, 5947 CrossRef PubMed.
- See the ESI‡ for details.
- (a) Y. Hatanaka and T. Hiyama, J. Org. Chem., 1989, 54, 268 CrossRef CAS; (b) H. F. Sore, W. R. J. D. Galloway and D. R. Spring, Chem. Soc. Rev., 2012, 41, 1845 RSC; (c) K. Hirabayashi, J. Kawashima, Y. Nishihara, A. Mori and T. Hiyama, Org. Lett., 1999, 1, 299 CrossRef CAS.
- For selected reviews on versatile transformations of 1,3-dienes, see: (a) J. Pyziak, J. Walkowiak and B. Marciniec, Chem. – Eur. J., 2017, 23, 3502 CrossRef CAS PubMed; (b) Y. Xiong, Y. Sun and G. Zhang, Tetrahedron Lett., 2018, 59, 347 CrossRef CAS; (c) X. Wu and L.-Z. Gong, Synthesis, 2019, 122 Search PubMed.
Footnotes |
| † Dedicated to Professor Koichi Narasaka with Gratitude on the Occasion of his 77th Birthday (Kiju). |
| ‡ Electronic supplementary information (ESI) available: Experimental procedures, characterization for new compounds including NMR spectra. See DOI: 10.1039/d1cc06761c |
| This journal is © The Royal Society of Chemistry 2022 |