
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAdvances in allylic and benzylic C–H bond functionalization enabled by metallaphotoredox catalysis
Huifeng
Yue†
 *a,
Chen
Zhu†
*a,
Long
Huang
b,
Abhishek
Dewanji
b and
Magnus
Rueping
*a
*a,
Chen
Zhu†
*a,
Long
Huang
b,
Abhishek
Dewanji
b and
Magnus
Rueping
*a
aKAUST Catalysis Center, KCC, King Abdullah University of Science and Technology, KAUST, Thuwal 23955-6900, Saudi Arabia. E-mail: magnus.rueping@kaust.edu.sa
bInstitute of Organic Chemistry, RWTH Aachen University, Landoltweg 1, 52074 Aachen, Germany
First published on 6th December 2021
Abstract
Metallaphoto-catalysis has been established as a robust platform for efficient construction of a range of chemical bonds. Moreover, transformation of native functionalities such as C(sp3)–H bonds to produce functional molecules represents one of the most attractive strategies in organic synthesis. Merging two powerful methodologies, metallaphoto-catalyzed benzylic and allylic C(sp3)–H bond functionalizations provide a series of general and mild approaches for diversification of alkylbenzenes and alkenes.
1. Introduction
Transformation of native functionalities to produce high value-added compounds has always been a critical goal pursued by chemists studying organic catalysis and synthesis. C–H bonds are among the most ubiquitous chemical bonds in nature; therefore, direct functionalization of such bonds is of great significance due to its step and atom economy. The last few decades have witnessed the development of methodologies for C(sp2)–H bond activation, and a wide range of valuable organic compounds have been accessed via these powerful advancements.1–9 Due to the abundance of starting materials such as hydrocarbons, greater emphasis has been placed on functionalization of C(sp3)–H bonds in recent years, and both C–C and C-heteroatom bond formation has been achieved via different catalytic approaches.10–17 Despite these advancements, a number of limitations remain, most notably associated with the use of precious metals with relatively high loadings or the need for directing groups, stoichiometric oxidants, or high temperatures.Over the past decade, photocatalysis has proven to be a powerful platform for a wide range of organic transformations that would otherwise be unavailable.18–25 In particular, photoredox-catalyzed C–H bond functionalization has been established, and it allows site-selective activation of inert alkanes.26–28 Further combination of photocatalysis with transition metals in C–H bond activation enabled a wide range of cross-coupling reactions using hydrocarbons as coupling partners.29–31 Given that the number of publications on metallaphoto-catalyzed allylic and benzylic C–H bond functionalization is rapidly increasing, this feature article will mainly focus on newly developed protocols in this area. In general, alkyl radicals can be delivered via hydrogen atom transfer (HAT) between the alkyl benzene/alkene and the excited photocatalyst, or HAT reagents. Direct oxidation of alkenes by excited photocatalysts and subsequent deprotonation could also afford allylic radicals. The resulting allylic or benzylic radical could then react with a variety of metal catalysts. These newly established protocols are categorized based on the types of products they generate: (1) arylation and alkenylation of benzylic and allylic C(sp3)–H bonds; (2) acylation of benzylic and allylic C(sp3)–H bonds; (3) alkylation of benzylic and allylic C(sp3)–H bonds; (4) carboxylation of benzylic and allylic C(sp3)–H bonds; (5) alkynylation of benzylic and allylic C(sp3)–H bonds; and (6) sulfonylation and azidation of benzylic and allylic C(sp3)–H bonds. We believe that this classification will help the community understand this type of reaction systematically and will be beneficial to further development of more diverse benzylic and allylic C(sp3)–H bond functionalization methodologies under mild conditions (Scheme 1).
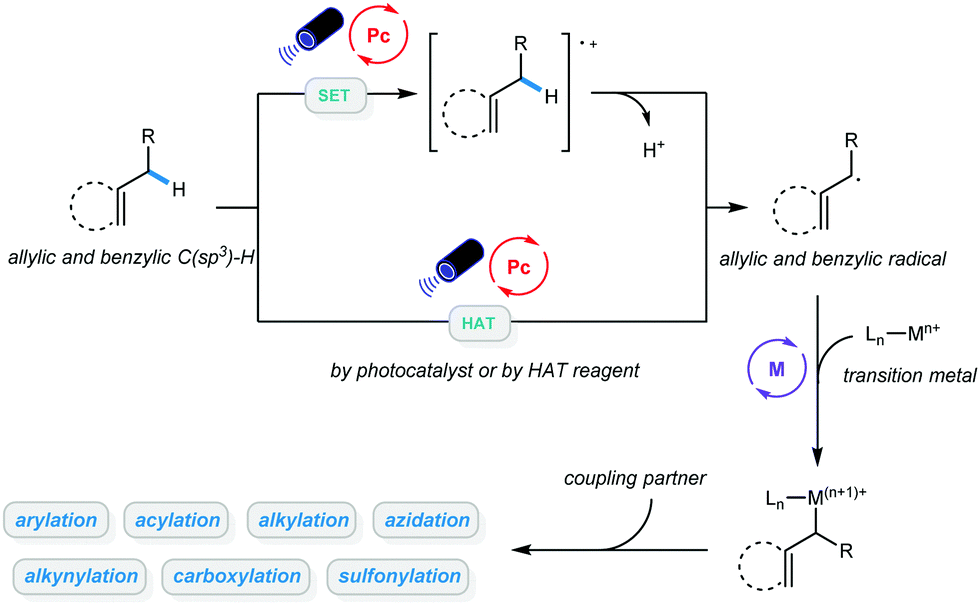 | ||
| Scheme 1 Metallaphoto-catalyzed allylic and benzylic C–H bond functionalization. | ||
2. Arylation and alkenylation of benzylic and allylic C(sp3)–H bonds
The construction of C(sp3)–C(sp2) bonds has long been a vital aspect of increasing molecular complexity. In this context, elegant studies on photoredox and nickel dual-catalyzed arylation of unactivated C(sp3)–H bonds were reported in 2016 by MacMillan, Doyle, and Molander, who realized C(sp3)–H activation of amines and ethers, respectively.32–34 For each protocol, one example of benzylic C(sp3)–H arylation of toluene was provided (Scheme 2).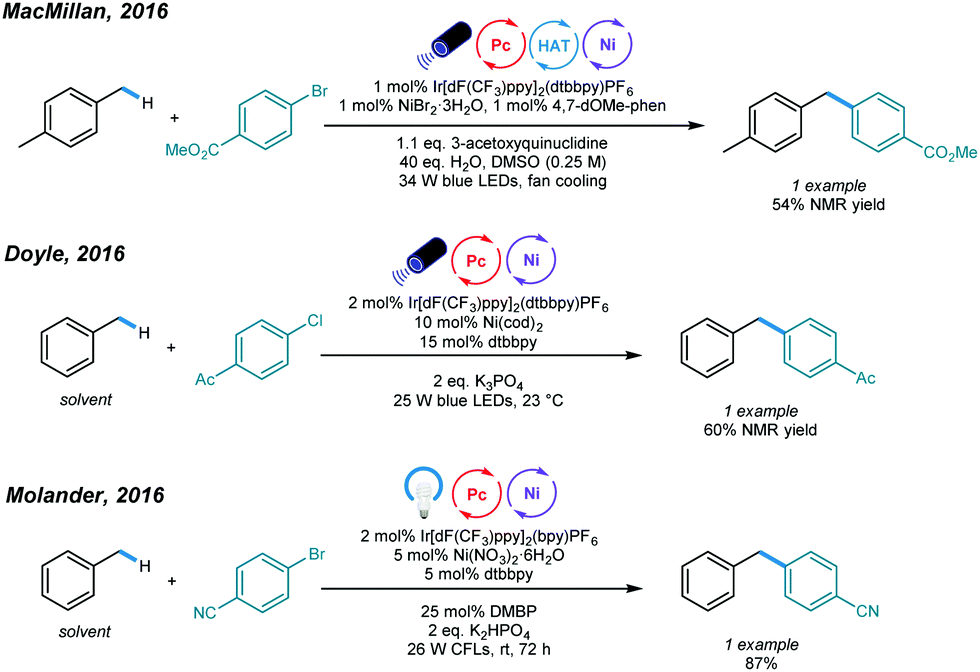 | ||
| Scheme 2 Photoredox and nickel dual-catalyzed arylations of unactivated C(sp3)–H bonds. | ||
One year later, an interesting UV light-induced benzylic C–H arylation of methylbenzenes via nickel catalysis was developed by Murakami and coworkers (Scheme 3).35 In this catalytic system, 5.0 mol% of NiCl2·dtbbpy complex and 365 nm UV light were used. The catalytic cycle starts with UV light-induced homolysis of NiCl2·dtbbpy, which generates the active NiI–X catalyst. Oxidative addition of an aryl bromide gives an Ar–NiIII(X)–Br intermediate that is transformed to [Ar–NiIII(X)–Br]* upon irradiation with UV light. Homolysis of the resulting NiIII species gives an Ar–NiII–Br intermediate and X˙ radical. The X˙ radical quickly abstracts one H atom to produce the benzyl radical that is trapped by Ar–NiII–Br to form a reactive Bn–NiIII(Br)–Ar intermediate. Reductive elimination from the unstable NiIII species delivers the product and regenerates the active NiI–X catalyst. UV-vis absorption spectra of the components rationalizes the proposed mechanism. The scope with regard to aryl bromides was broad, as illustrated by tolerance of electron-withdrawing and electron-donating groups, including trifluoromethyl, fluoro, chloro, ester, cyano, pyridine, thiophene, and even reactive boronic esters. A series of toluene derivatives served as feasible coupling partners. Additionally, tribenzylbenzene was obtained from tribromobenzene in a single operation with a high yield.
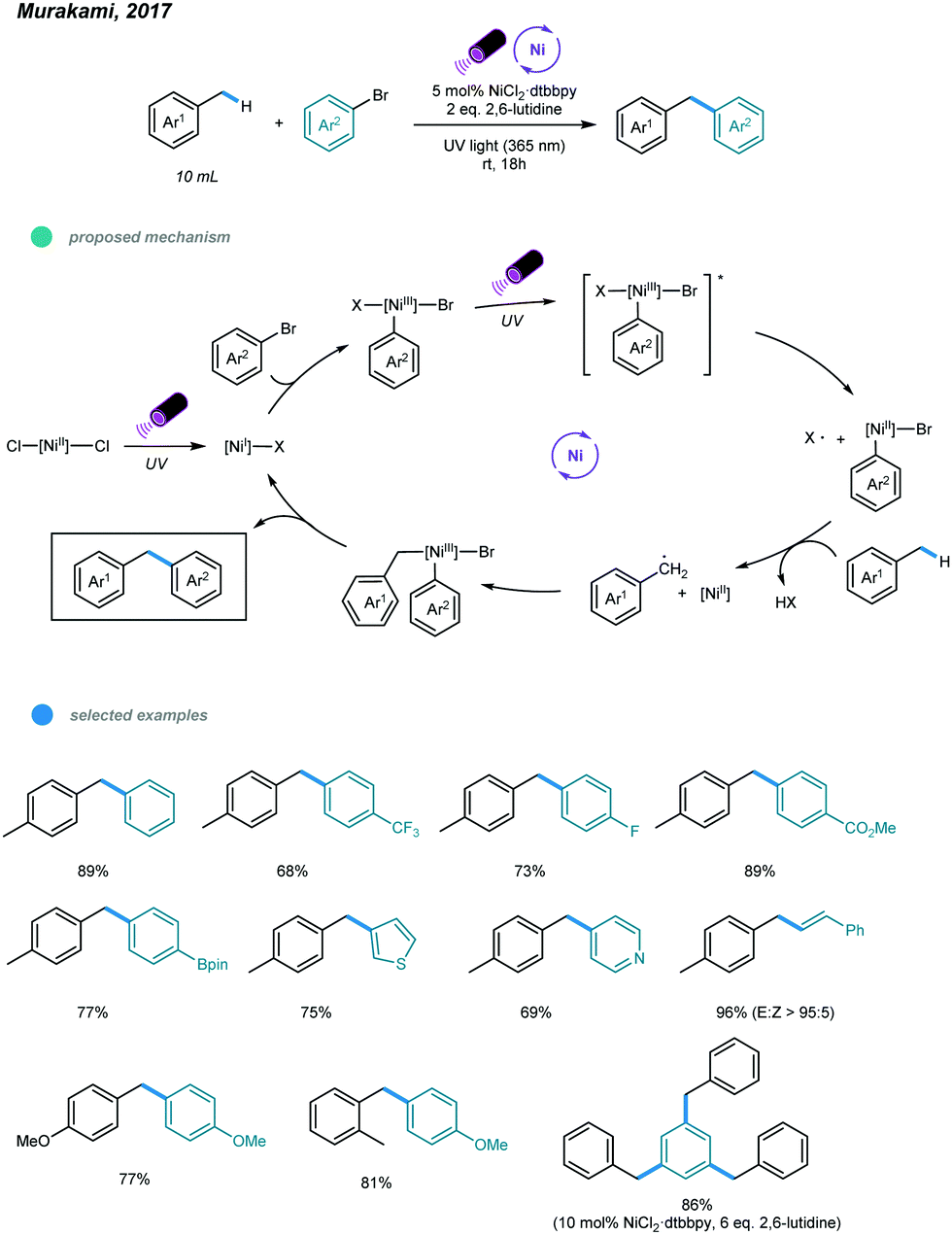 | ||
| Scheme 3 UV light-induced nickel-catalyzed benzylic C–H arylations of methylbenzenes. | ||
Although arylation of benzylic C(sp3)–H bonds has been realized, arylation of allylic C(sp3)–H bonds remained undeveloped. To address this issue, Rueping and coworkers developed a photoredox and nickel dual-catalyzed protocol realizing cross-coupling of allylic C(sp3)–H bonds with aryl and vinyl bromides in 2018 (Scheme 4).36 A series of highly valuable allylarenes and 1,4-dienes were obtained from unactivated alkenes under mild conditions. The methodology features the first combination of an inexpensive acridinium photocatalyst and a nickel-metal catalyst. Mechanistically, irradiation with blue light excites the photocatalyst Mes–Acr–Me+ to give excited [Mes–Acr–Me+]*, which undergoes triplet–triplet energy transfer with Ar–NiII–Br to form an electronically excited [Ar–NiII–Br]* species. Then, the excited NiII species undergoes homolysis to give a bromine radical that abstracts a hydrogen atom from the allylic C(sp3)–H bond of an olefin, generating an allylic radical. Addition of the allylic radical to the active NiI species affords a Ar–NiII–allylic intermediate, and reductive elimination delivers the coupled product. Radical trapping experiments with TEMPO and UV light control experiments were carried out and support this mechanism. A wide range of electron-deficient aryl bromides and heterocyclic substrates worked well in this allylation system, giving the corresponding products in moderate to excellent yields. Additionally, vinyl bromides reacted smoothly with alkenes, providing an efficient way to construct valuable 1,4-dienes with good stereoselectivity. In addition, a series of tri- and tetrasubstituted alkenes participated and provided moderate to good yields. Notably, the reactions exhibit excellent regioselectivities, and coupling tends to replace primary C–H bonds rather than methylene and methine C–H bonds. Additionally, monoarylation adducts were observed exclusively, which may be due to steric effects. The problems remaining to be addressed are the use of electron-rich aryl bromides and achieving Z/E control when using asymmetric alkenes as coupling partners.
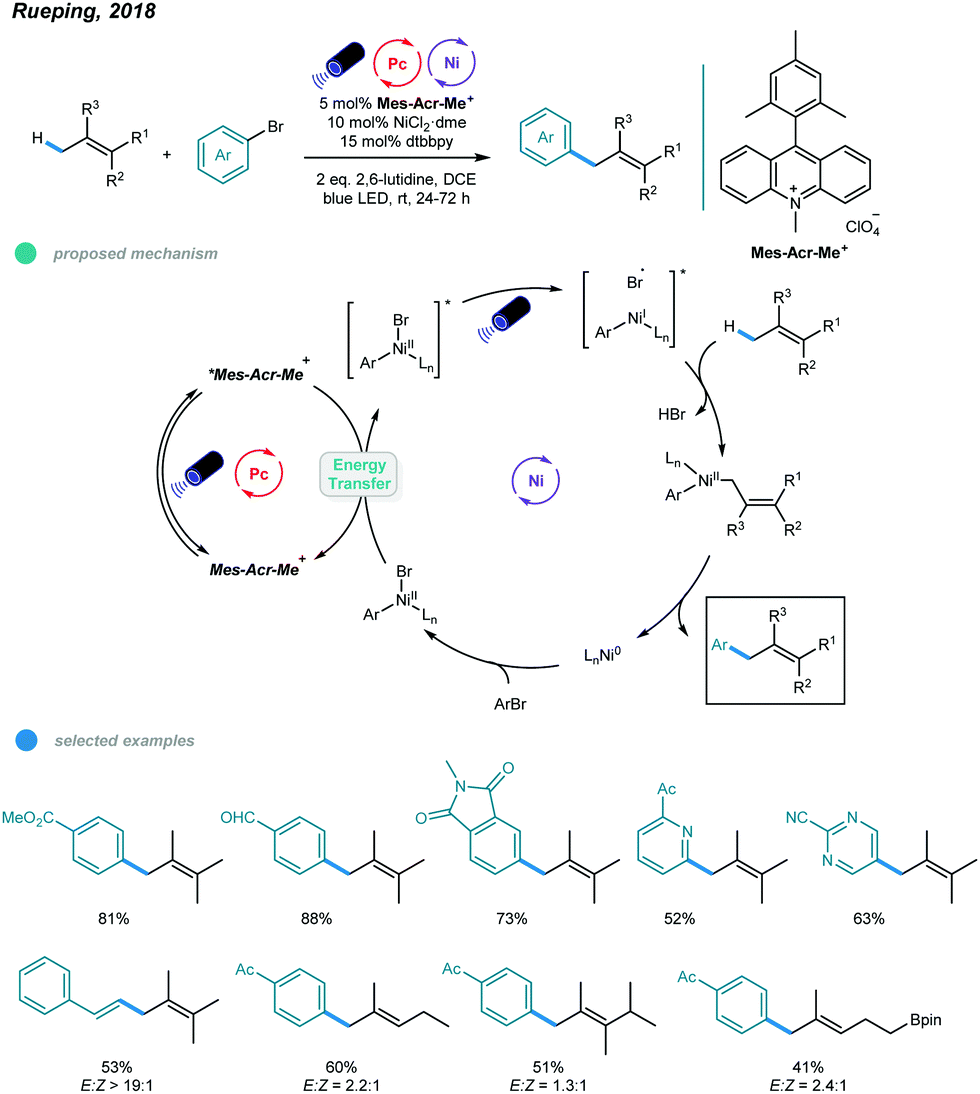 | ||
| Scheme 4 Photoredox and nickel dual-catalyzed allylic C(sp3)–H arylations and vinylations. | ||
Making use of a diaryl ketone photosensitizer, Rueping and coworkers later extended the arylation scope to benzylic C(sp3)–H bonds and established a photo/nickel dual catalyzed method for direct arylation of toluene derivatives (Scheme 5).37 The use of benzophenone enables generation of benzylic radicals from toluene, and the resulting benzylic radicals then participate in the nickel catalytic cycle and form a series of diarylmethanes. As to the mechanism, photoexcitation of the diaryl ketone photosensitizer PS produces the excited triplet state PS*, which abstracts a H atom from toluene and gives a benzyl radical that subsequently adds to the active Ni0 species to form a Bn–NiI intermediate. Oxidative addition of an aryl halide to the resulting NiI species leads to formation of an unstable Bn–NiIIIBr–Ar intermediate that tends to undergo reductive elimination to deliver the coupling product and NiI–Br. Reduction of this NiI-Br by reduced PS (PS–H) with the aid of a base regenerates the active Ni0 catalyst and completes the catalytic cycle. An array of aryl bromides bearing electron-withdrawing and electron-donating groups reacted with toluene smoothly, affording the corresponding diarylmethanes in moderate to excellent yields.
 | ||
| Scheme 5 Photoredox and nickel dual-catalyzed benzylic C(sp3)–H arylations of toluene derivatives. | ||
Substrates with fused rings and heterocycles also underwent this transformation and showed good yields. The reaction occurred at the C–Br bond for 4-chlorobromobenzene, and the bromotolyl benzylic radical was not observed, indicating good chemoselectivity for this benzylic arylation. Notably, steric hindrance has little influence on reaction activity. In addition, electron-deficient and heterocyclic aryl chlorides were also suitable for this transformation. Xylenes and mesitylene also showed excellent reactivity. When applying the standard reaction conditions to aryl iodides, the reactivity was much lower. Interestingly, the addition of one equivalent of tetrabutyl ammonium bromide (TBAB) as an additive improved the yields significantly. These results, together with those of control experiments, indicate that the diaryl ketone photosensitizer acts as both a hydrogen atom transfer agent and an energy transfer agent. Moreover, scale-up was performed and proceeded with high yield. The use of alkylbenzenes other than methyl benzenes seems ineffective in this protocol. It is noted that the MacMillan and Martin groups also independently reported nickellaphoto-catalyzed arylation of C(sp3)–H bonds, and three examples of alkylbenzenes were presented (Scheme 5).38,39
Very recently, Lee and coworkers realized the benzylic C(sp3)–H arylations of indole derivatives (Scheme 6).40 The facile oxidation of indoles enabled the efficient generation of the benzylic radical via a sequential electron transfer–proton transfer (ET/PT). Particularly, the radical cation was first generated via the oxidation of the indole by the excited Ir-based photocatalyst, followed by the deprotonation to give the benzylic radical. The resulting benzylic radical was trapped by the active Ni0 catalyst to deliver a NiI complex, which undergoes oxidative addition with aryl bromide to afford a NiIII complex. Next, the unstable NiIII complex undergoes facile reductive elimination to furnish the arylation product and NiI–Br species that is reduced to regenerate the active Ni0 catalyst. The scope regarding to both indole substrates and aryl bromides are broad with even complex drug molecule derivatives applicable. However, other types of heterocycles including benzofuran, thiophene and pyrrole failed to give to desired products.
 | ||
| Scheme 6 Photoredox and nickel dual-catalyzed benzylic C(sp3)–H arylation of indole derivatives. | ||
Although these protocols provide efficient ways to realize functionalization of allylic or benzylic C–H bonds, they are limited to achiral processes, which may be due to the lack of efficient chiral ligands. To address this issue, Lu and coworkers designed a series of chiral biimidazoline (BiIM) ligands and realized enantioselective benzylic C(sp3)–H arylation of alkylbenzenes with aryl bromides (Scheme 7).41 Similarly, oxidative addition of aryl halide to the Ni0 species generated in situ provides an Ar–NiII–Br complex, which is oxidized by the excited *IrIII catalyst to give a new Ar–NiII species and Br˙ radical. The Br˙ radical abstracts a hydrogen atom from the alkylbenzene to generate a benzylic radical that subsequently adds to the Ar–NiII species to afford a NiIII complex. Then, the chiral 1,1-diaryl alkane product is produced via reductive elimination from the NiIII complex, which also yields a NiI intermediate. SET occurs between the NiI intermediate and the reducing IrII species, regenerating the Ni0 species and the ground state IrIII photocatalyst. After optimization, the authors found that addition of bis(4-methoxyphenyl)methanone (DMBP) as a cophotocatalyst improved the yield, and the use of sterically hindered N-3-tBuPh-iPrBiIM gave the highest yield and ee value. Electron-rich and electron-deficient aryl bromides, as well as polycyclic and heterocyclic aryl bromides, all afforded the corresponding chiral 1,1-diaryl alkanes in moderate to good yields and with good enantioselectivity. A number of substituted benzenes (4 equivalents) underwent this asymmetric benzylic C–H arylation, and moderate yields with good ers were observed after 96 h. It should be noted that only secondary benzylic C–H bonds could be arylated, and tertiary benzylic C–H bonds were unreactive in this protocol. Later, the same group also developed the photoredox and nickel dual catalyzed stereo- and enantioselective benzylic C–H alkenylation (Scheme 7).42 A wide range of chiral allylic compounds were obtained in up to 93% ee and >20/1 E/Z ratio. The high stereoselectivity attributes to the skillful use of stilbene as a triplet-energy inhibitor, which declines photocatalytic E/Z isomerization of the product.
 | ||
| Scheme 7 Photoredox and nickel dual-catalyzed enantioselective benzylic C(sp3)–H arylation of alkylbenzenes. | ||
3. Acylation of benzylic C(sp3)–H bonds
Ketones and esters are two of the most important and useful classes of compounds in organic chemistry. Accordingly, efficient methods for preparation of these functional groups are always desirable. In 2018, the Doyle group developed a photoredox and nickel dual-catalyzed protocol for acylation of unactivated C–H bonds in alkanes and toluene derivatives (Scheme 8).43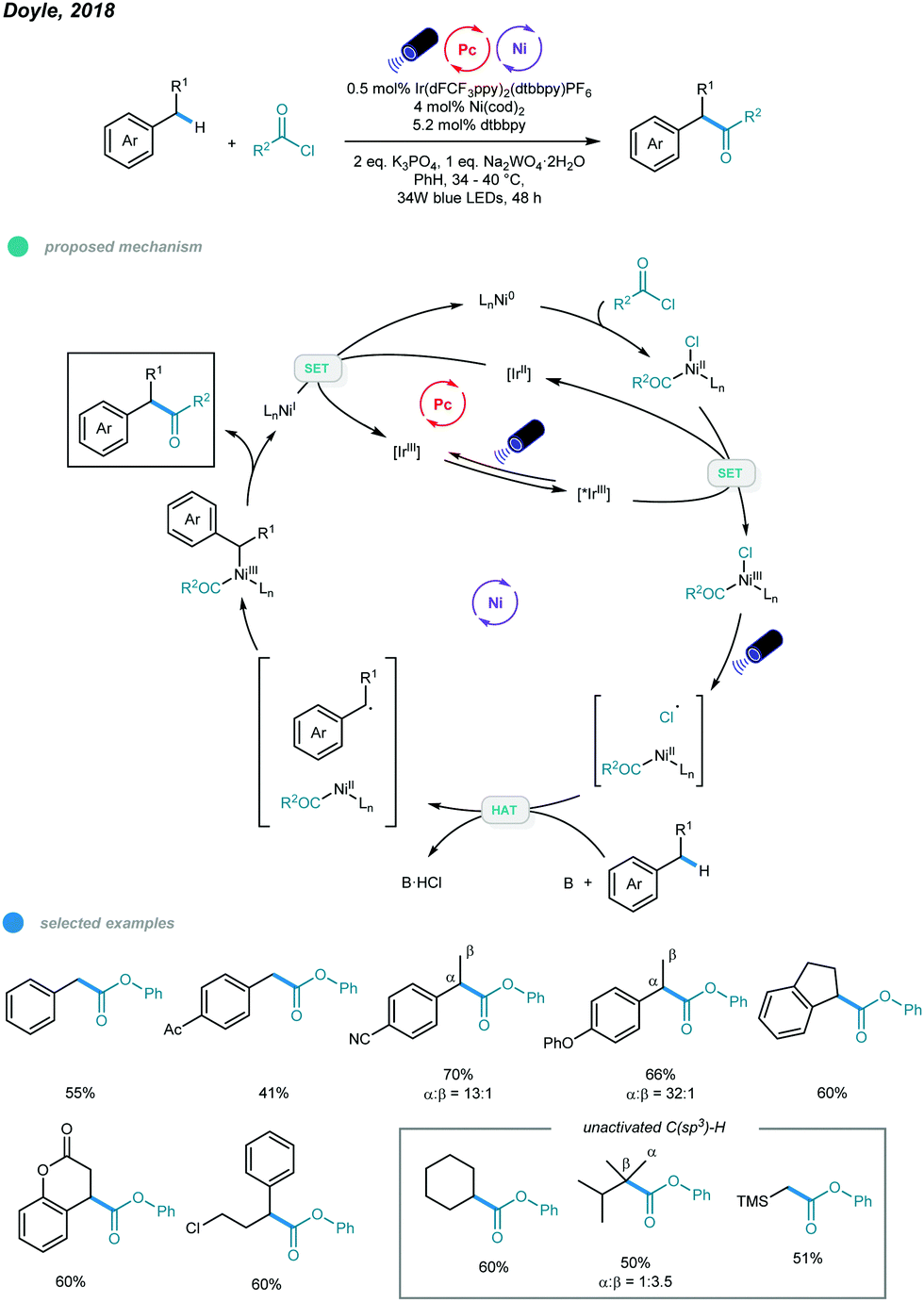 | ||
| Scheme 8 Photoredox and nickel dual-catalyzed acylation of unactivated C–H bonds of alkanes and toluene derivatives. | ||
Various carbonyl derivatives were obtained via site-selective cross-coupling of acyl chlorides and hydrocarbons. The reaction starts with oxidative addition of a chloroformate derivative to Ni0, generating a NiII(CO2R)(Cl) complex that is then oxidized by the excited *IrIII photocatalyst to form a NiIII intermediate. The Cl˙ radical is then generated via photoelimination from the NiIII intermediate upon irradiation with blue light. Hydrogen atom abstraction of hydrocarbons by Cl˙ radicals leads to a carbon-centered radical that was trapped by the NiII intermediate to afford an unstable NiIII intermediate. The acylation product is produced via reductive elimination from the resulting NiIII intermediate and a NiI intermediate is generated. Finally, the active Ni0 catalyst is regenerated via reduction of the NiI intermediate by the reducing IrII photocatalyst. Notably, in addition to toluene, other acyclic and cyclic alkyl benzenes all gave the acylated product with good efficiency and regioselectivity. The utilization of air-sensitive Ni(cod)2 necessitates further improvement.
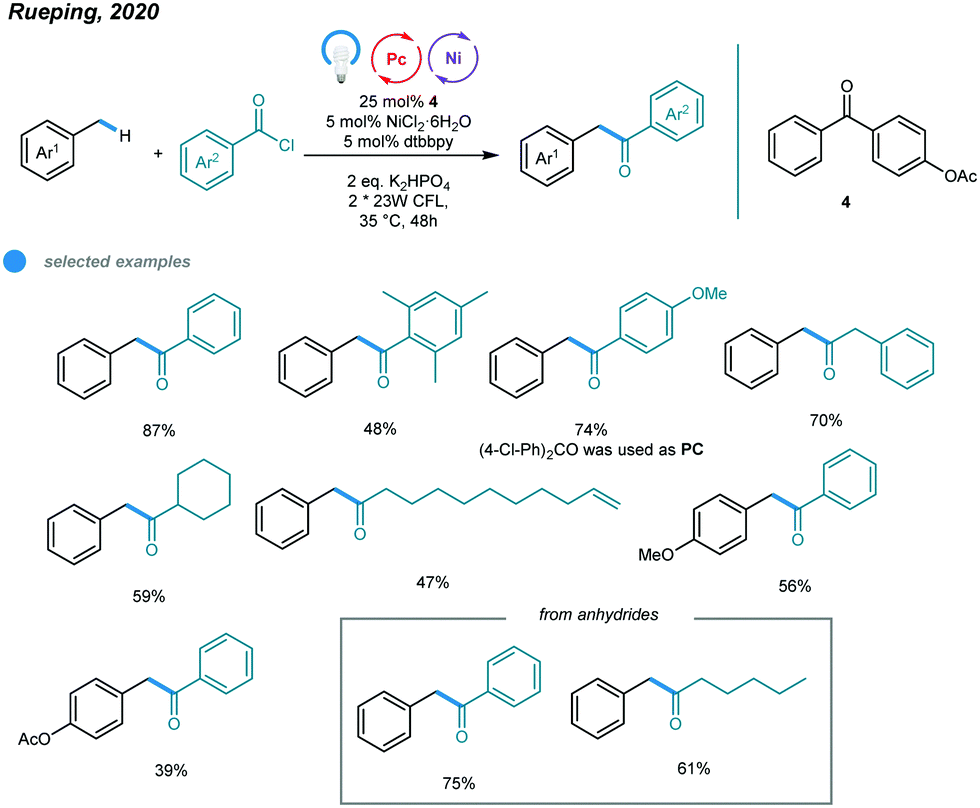
Another methodology for the synthesis of unsymmetrical ketones was developed by Rueping and coworkers. They realized a photoinduced nickel-catalyzed benzylic C–H acylation reaction using 4-benzoylphenyl acetate as a photosensitizer (Scheme 9).44 A series of unsymmetrical ketones were prepared via cross-coupling of methylbenzenes with acid chlorides and anhydrides. The mechanism is similar to that for benzylic C–H arylation. Both aromatic and aliphatic acid chlorides were successfully converted to the corresponding ketones with moderate to high efficiency. The steric hindrance of methylbenzene significantly affected the reactivity of the transformation, as illustrated by the fact that ortho-xylene furnished the acylation product in low yield. In addition, benzoic anhydrides, caproic anhydride and pivalic anhydride were all effective acylating reagents. This method provides a general and attractive alternative with which to access valuable ketones.
 | ||
| Scheme 9 Photoredox and nickel dual-catalyzed acylation of benzylic C–H bonds of toluene derivatives. | ||
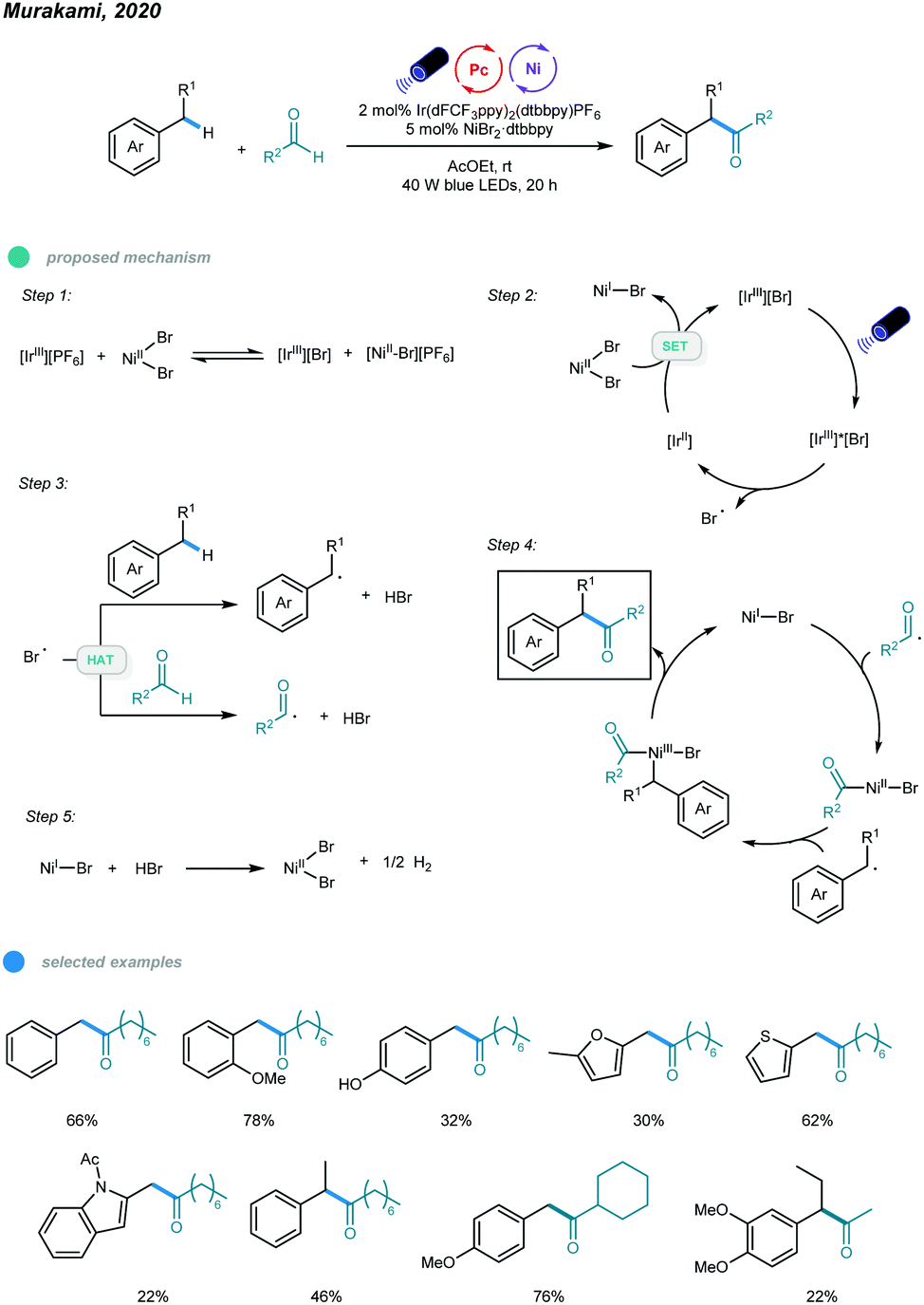
In 2020, Murakami, Ishida, and coworkers realized photoredox and nickel dual-catalyzed acylation of alkylbenzenes (Scheme 10).45 A number of α-aryl ketones were prepared via a dehydrogenative coupling reaction between benzylic and aldehydic C–H bonds. Mechanistically, IrIIIPF6 first undergoes anion exchange with NiBr2 to form an IrIIIBr complex. This IrIIIBr complex is excited upon irradiation with blue LEDs and then undergoes SET from bromide anion to iridium(III) to afford a Br˙ radical and the IrII species that reduces NiBr2 to a NiI–Br species. Abstraction of benzylic and aldehydic H atoms by the resulting Br˙ radical generates benzylic and acyl radical species, respectively. Addition of the two radicals to NiI–Br species forms Bn–NiIIIBr–acyl intermediates, which deliver the α-aryl ketone product via reductive elimination. Finally, the NiBr2 catalyst is regenerated by reaction of the NiI–Br species and HBr with the release of H2 gas. Electron-rich toluene derivatives, as well as methyl groups containing thiophene, furan, and indole, coupled smoothly with the aldehyde.
 | ||
| Scheme 10 Photoredox and nickel dual-catalyzed acylation of C–H bonds of alkylbenzenes with aldehydes. | ||
Notably, the challenging substrate ethylbenzene also worked with this protocol and provided the corresponding product in moderate yield. Cyclic, linear and α-branched aliphatic aldehydes were successfully applied. However, toluene derivatives bearing electron-withdrawing groups such as alkoxycarbonyl, acyl, and cyano, as well as aromatic aldehydes, gave no coupling products.
It is noted that the efficient benzylic C–H acylation of indoles has been realized by the Lee group using symmetric acid anhydrides as the acyl sources.40
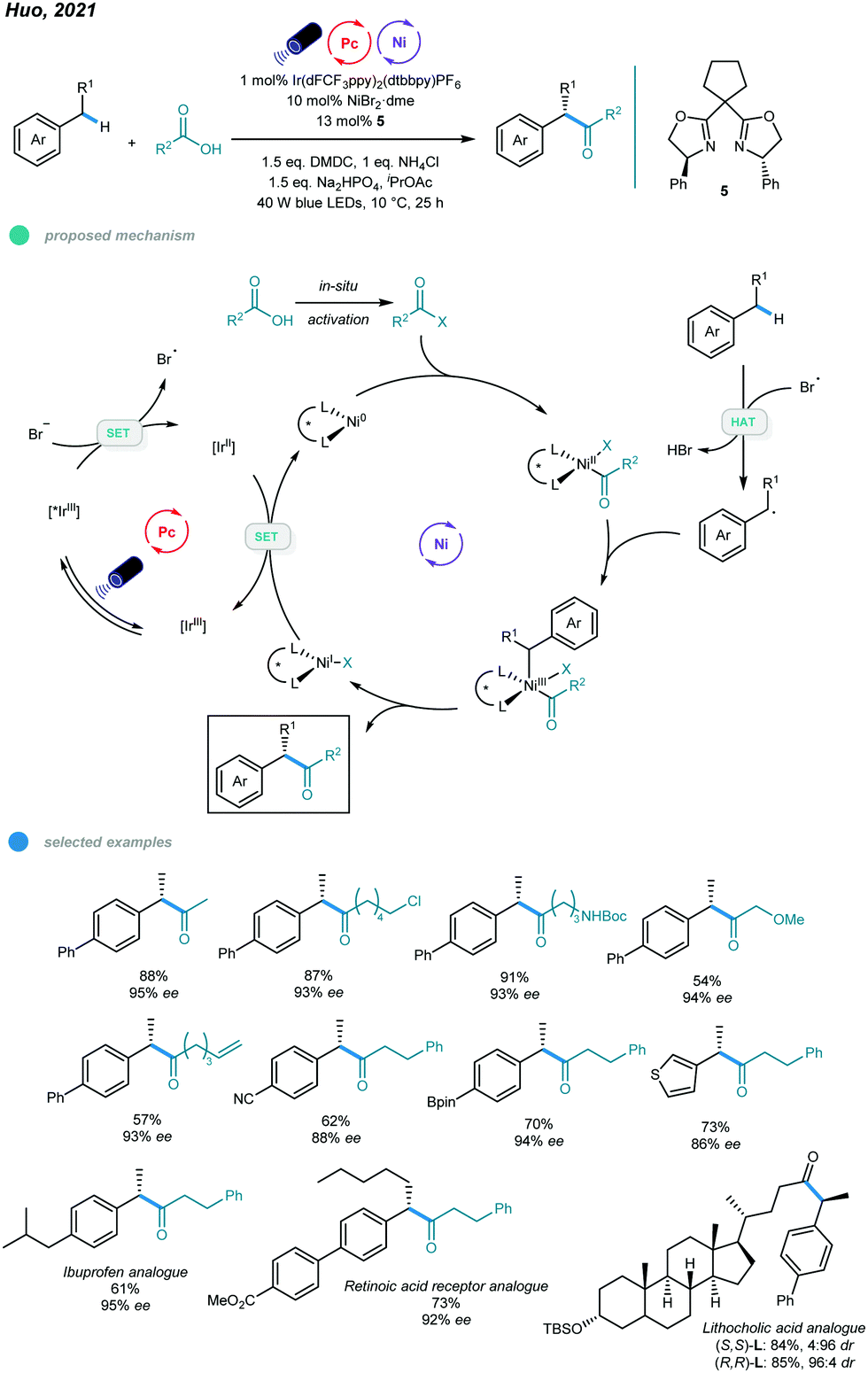
More recently, an efficient and versatile methodology for asymmetric acylation of benzylic C–H bond was established by Huo and coworkers (Scheme 11).46 Photoredox and nickel dual-catalyzed cross-coupling reactions of feedstock carboxylic acids and simple alkyl benzenes provide straightforward access to α-aryl ketones and α-aryl esters with high enantioselectivities. In the mechanism, the bromide anion is initially oxidized to the bromine radical via SET with an excited *IrIII photocatalyst, which forms a reducing IrII species. The bromine radical abstracts a hydrogen atom from the alkylbenzene to give a benzylic radical. Additionally, the carboxylic acid derivative undergoes oxidative addition with the active Ni0 catalyst to form an acyl–NiII intermediate, which traps the resulting benzylic radical to furnish an acyl–NiIII–Bn intermediate. Reductive elimination from the NiIII intermediate delivers the desired product and a NiI species. Finally, reduction of the NiI species by the reducing IrII species regenerates the active Ni0 species and completes the catalytic cycles. Reaction screening showed that the use of NiBr2·glyme as a nickel source and chiral bis(oxazoline) as a ligand ensures high reactivity and enantioselectivity. The scope with regard to both carboxylic acids and alkylarenes is broad, and various functional groups, such as chloride, bromide, olefin, boronate ester, and heterocycles, were tolerated. Moreover, late-stage functionalizations of a series of pharmaceutically relevant molecules were carried out, yielding the corresponding drug analogs with moderate to good yields and high enantioselectivities. Notably, aromatic carboxylic acids participated in this reaction and gave only modest yields and enantioselectivities, and the sterically hindered coupling partners also participated inefficiently, giving low yields or no product.
 | ||
| Scheme 11 Photoredox and nickel dual-catalyzed asymmetric acylations of benzylic C–H bonds with carboxylic acids. | ||
4. Alkylation of benzylic and allylic C(sp3)–H bonds
The Nozaki–Hiyama–Kishi (NHK) reaction represents one of the most powerful methods for allylation of aldehydes, but inherent drawbacks, such as stoichiometric generation of chromium waste or use of excess manganese, limit its application.47–49 In 2018, Glorius's group reported the first photoredox- and chromium-catalyzed allylic C–H functionalization protocol for allylation of aldehydes (Scheme 12).50 A wide array of homoallylic alcohols was synthesized with excellent diastereoselectivities via the reaction of aldehydes with electron-rich alkenes, including allyl (hetero)arenes, β-alkyl styrenes, and allyldiarylamines. In the mechanism, photoexcitation of Ir-based photocatalysts first gives excited state *IrIII, which oxidizes the electron-rich alkene to a radical cation intermediate; the radical cation is deprotonated to furnish an allylic radical. The resulting allylic radical is then trapped by the CrII catalyst to form an allyl CrIII intermediate, which subsequently reacts with aldehyde via a six-membered transition state to generate chromium alkoxide in an anti-selective manner. Protonolysis of chromium alkoxide delivers the homoallylic alcohol product along with a new high-valent CrIII intermediate. Reduction of this CrIII complex by the reducing IrII photocatalyst regenerates the active CrII catalyst and completes the catalytic cycle. Mechanistic studies, including chromium-free reactions, Stern–Volmer quenching, radical trapping, and ring-opening control experiments, suggest the formation of organochromium intermediates and support the proposed mechanism.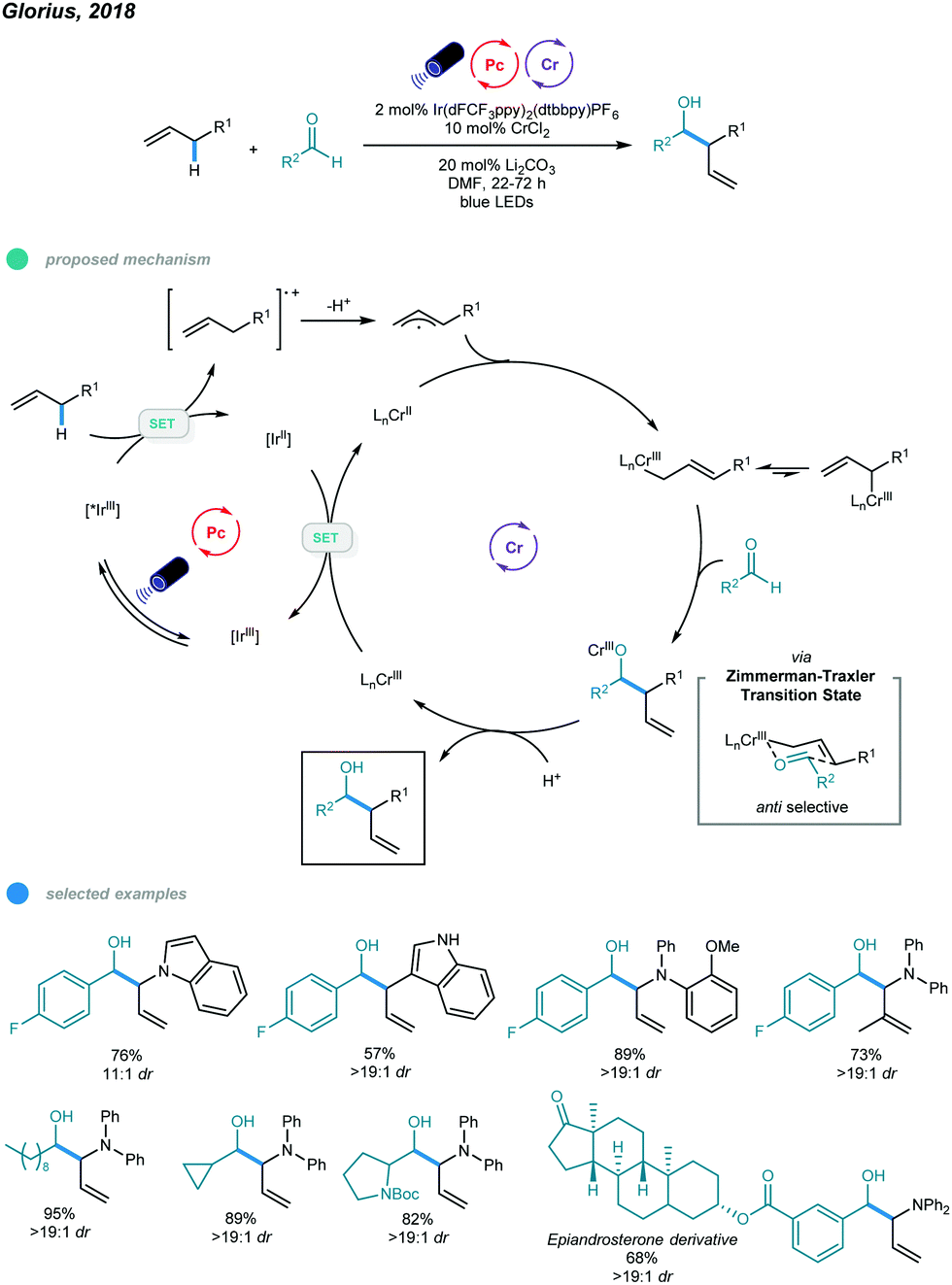 | ||
| Scheme 12 Photoredox and chromium dual-catalyzed allylations of aldehydes. | ||
A series of allyl-indoles and N-allyl-carbazoles, including those bearing free N–H bonds, worked well and gave the corresponding products in high yields with excellent diastereoselectivities. Other alkenes, such as electron-rich allylic benzenes, β-alkyl styrenes, and N-allyl-diarylamines, were also used in this allylation protocol and exhibited good efficiency. However, less electron-rich allylic benzenes afforded the C–H alkoxylated product in low yield despite the utilization of a more oxidizing photocatalyst. Linear, branched, and cyclic aliphatic aldehydes, as well as aromatic aldehydes bearing reactive halides and bulky ortho-substituents, all underwent this transformation and showed good yields with exclusive diastereoselectivities. Interestingly, an epiandrosterone derivative containing other types of carbonyl moieties reacted with N-allyl-diphenylamine with high chemoselectivity. Additionally, a scaled-up reaction was successfully performed and gave a high yield. However, although primary and secondary aliphatic aldehydes participated well in this allylation reaction, tertiary aliphatic aldehydes were ineffective substrates, probably due to steric effects.
One year later, asymmetric allylation of aldehydes was realized by Kanai and coworkers via photoredox and chromium dual-catalyzed allylic C(sp3)–H functionalization of unactivated alkenes (Scheme 13).51 A range of homoallylic alcohols was formed with excellent diastereoselectivities and enantioselectivities. The success of this protocol relies on generation of organometallic intermediates from hydrocarbon alkenes. Specifically, allyl radicals, generated from oxidation of alkenes by excited organophotoredox acridinium catalysts and subsequent deprotonation of the generated cation radicals, are trapped by the chiral chromium(II) catalyst to give a chiral allyl chromium(III) complex. The resulting chromium(III) complex undergoes nucleophilic attack with an aldehyde in a syn-selective fashion, forming an enantiomerically enriched chromium alkoxide that provides the desired chiral homoallylic alcohol after protonolysis. Reduction of the oxidized chromium(III) complex regenerates the active chiral chromium(II) species. Interestingly, the addition of Mg(ClO4)2 as an additive significantly improves the reactivity and enantioselectivity, which may be due to its ability to enhance the radical cation concentration. Cyclic alkenes with different ring sizes and tri- and tetrasubstituted linear alkenes all reacted with aldehydes to give the corresponding products in moderate to high yield with up to 99% ees. Aromatic aldehydes bearing electron-neutral and electron-donating functional groups, heteroaryl aldehydes, and cyclic and linear aliphatic aldehydes were all feasible substrates, delivering chiral alcohols with good to excellent yields and enantioselectivities. Notably, 10 mol% CrCl3·3THF and 30 mol% NaOtBu were used instead of CrCl3·3THF for less reactive aldehydes, such as o-tolualdehyde and p-methoxybenzaldehyde, to obtain higher yields.
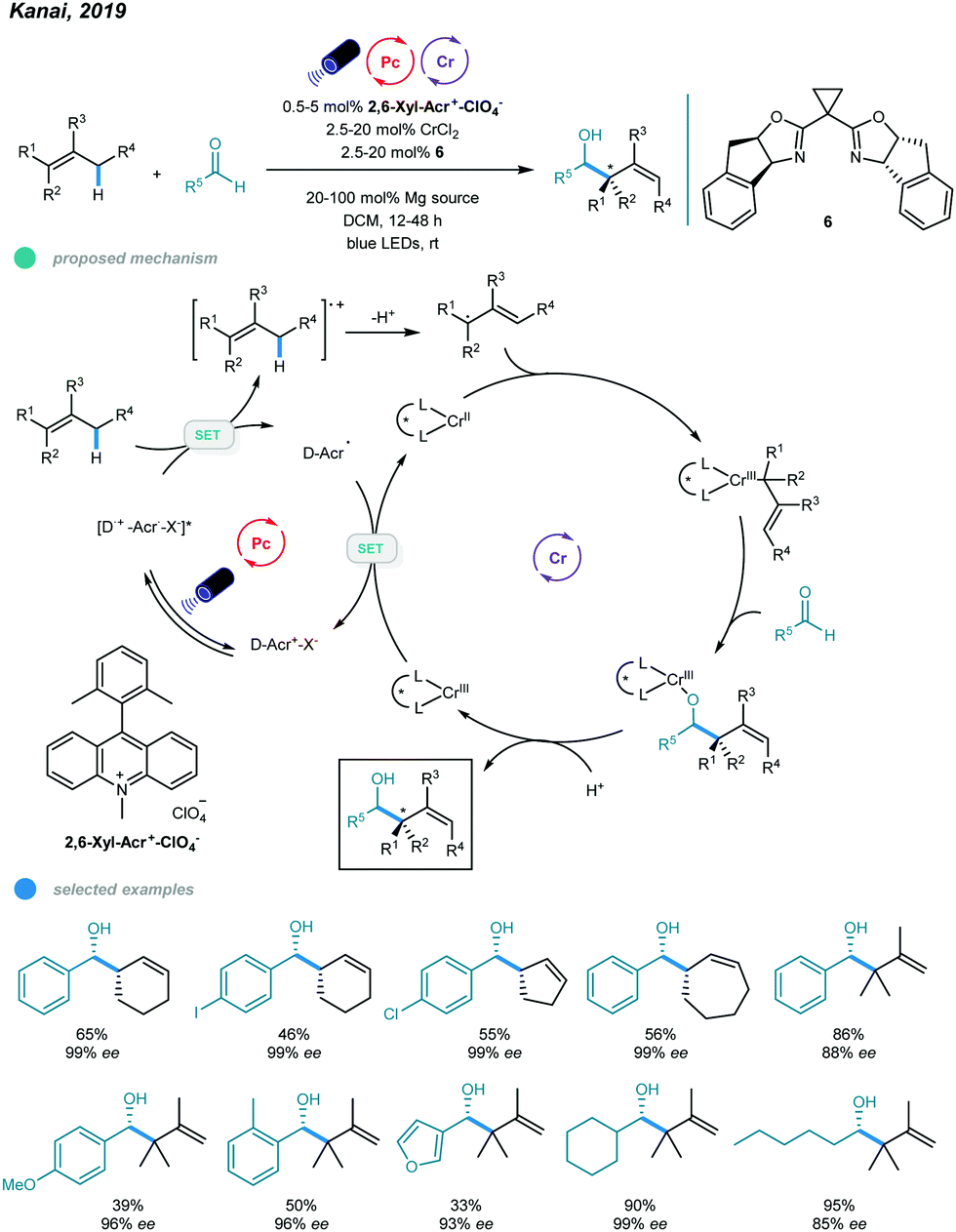 | ||
| Scheme 13 Photoredox and chromium dual-catalyzed asymmetric allylations of aldehydes. | ||
Although this protocol provides an efficient method for allylic functionalization of cyclic and tri- and tetrasubstituted alkenes, linear terminal alkenes and disubstituted internal alkenes such as 1-hexene and 2-butene are not functionalized. This limited scope is likely caused by their high oxidation potentials, which makes them difficult to oxidize by excited photoredox catalysts. To overcome this limitation, Kanai's group developed in 2020 a ternary hybrid catalyzed system comprising a photoredox catalyst, a hydrogen atom transfer (HAT) catalyst, and a chromium complex catalyst and realized catalytic allylation of aldehydes with even simple alkenes and structurally elaborated aldehydes (Scheme 14).52
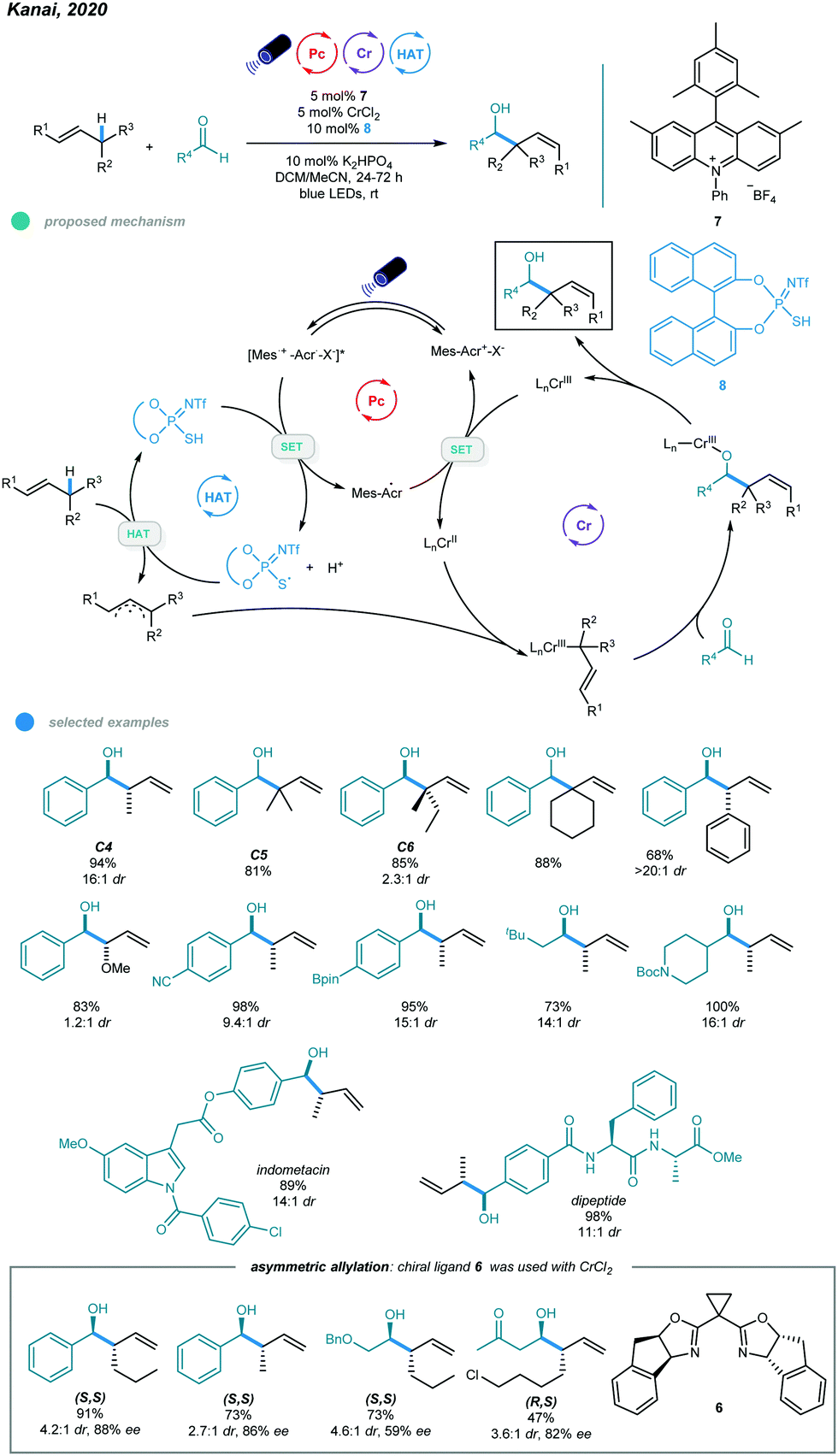 | ||
| Scheme 14 Photoredox and chromium dual-catalyzed allylation of aldehydes using unactivated alkenes. | ||
Unlike the previous report in which the allyl radical was generated via direct oxidation of the alkene and subsequent deprotonation, the allyl radical in this process was formed via the HAT pathway. In particular, initial oxidation of the thiophosphoric imide (TPI) HAT catalyst by excited acridinium catalyst gives a sulfur-centered radical RS˙ that abstracts a hydrogen atom from the allylic C–H bond of the alkene to afford the allyl radical. The subsequently formed allylchromium(III) species then attacks aldehydes via a six-membered chair transition state, furnishing the chromium alkoxides in an anti-selective manner. Finally, the homoallylic alcohol product is obtained after protonolysis. After screening, the authors found that the optimal reaction condition involved a combination of 5 mol% CrCl2, 10 mol% TPI as the HAT catalyst, 5 mol% acridinium salt as the photocatalyst, and 10 mol% K2HPO4 as the base in a mixed DCM and MeCN solvent system under blue LED irradiation at room temperature for 24 h. Most linear and branched alkenes, including C4, C5, C6 feedstocks, and some internal alkenes, reacted with aldehydes in good to excellent yields with high branch- and anti-selectivity, which was probably caused by a six-membered chair transition state for the (E)-2-hexenylchromium species. Although allyl ethers also participated in this reaction and exhibited high reactivity, the diastereoselectivity was low. The scope of aldehydes was quite broad, as illustrated by the use of aromatic and aliphatic aldehydes bearing steric and potentially sensitive functional groups and densely functionalized aldehydes, all of which participated in this process and exhibited good efficiency and diastereoselectivity. Notably, the transformation proceeded with site selectively to provide alkene C–H functionalization and leave the allylic and benzylic C–H bonds of aldehydes untouched. More importantly, the use of an indane-BOX ligand enabled catalytic asymmetric allylation of aldehydes with excellent regioselectivity, albeit with moderate to good diastereoselectivity and enantioselectivity.
In addition to photoredox and chromium dual catalysis, a combination of photoredox agents and other metals, such as copper, was also developed for alkylation of benzylic C(sp3)–H bonds. In 2018, Wu's group reported photoredox and copper-catalyzed reactions of alkylbenzenes and enones (Scheme 15).53 This Giese reaction between benzylic C–Hs and enones provides a convenient and efficient way to access value-added γ-aryl ketones. In this process, the benzylic radical is generated via oxidation of toluene by the excited state acridinium photocatalyst and subsequent deprotonation. Nucleophilic addition of the resulting benzylic radical to the Cu(OTf)2-activated enone gives a Lewis acid alkyl radical adduct, which is then reduced by the intermediate from the acridinium catalyst to form the corresponding cation intermediate. Finally, the cation intermediate undergoes protonation to deliver the Giese adduct. The scope of enones is extremely broad, as illustrated by the fact that electron-rich and electron-deficient benzylideneacetone, β-alkylated enones, simple vinyl ketones, trisubstituted benzylideneacetones, chalcone-type enones, aryl ketones bearing heterocycles, cyclic enones bearing different ring sizes, and unsaturated esters can all be employed to furnish benzylic conjugate addition products. Methylbenzenes containing steric ortho-methyl substituents, boronate esters, or pyridine as well as ethylbenzene and indane underwent this transformation smoothly in the presence of the dual catalysts, affording the desired products in good yields.
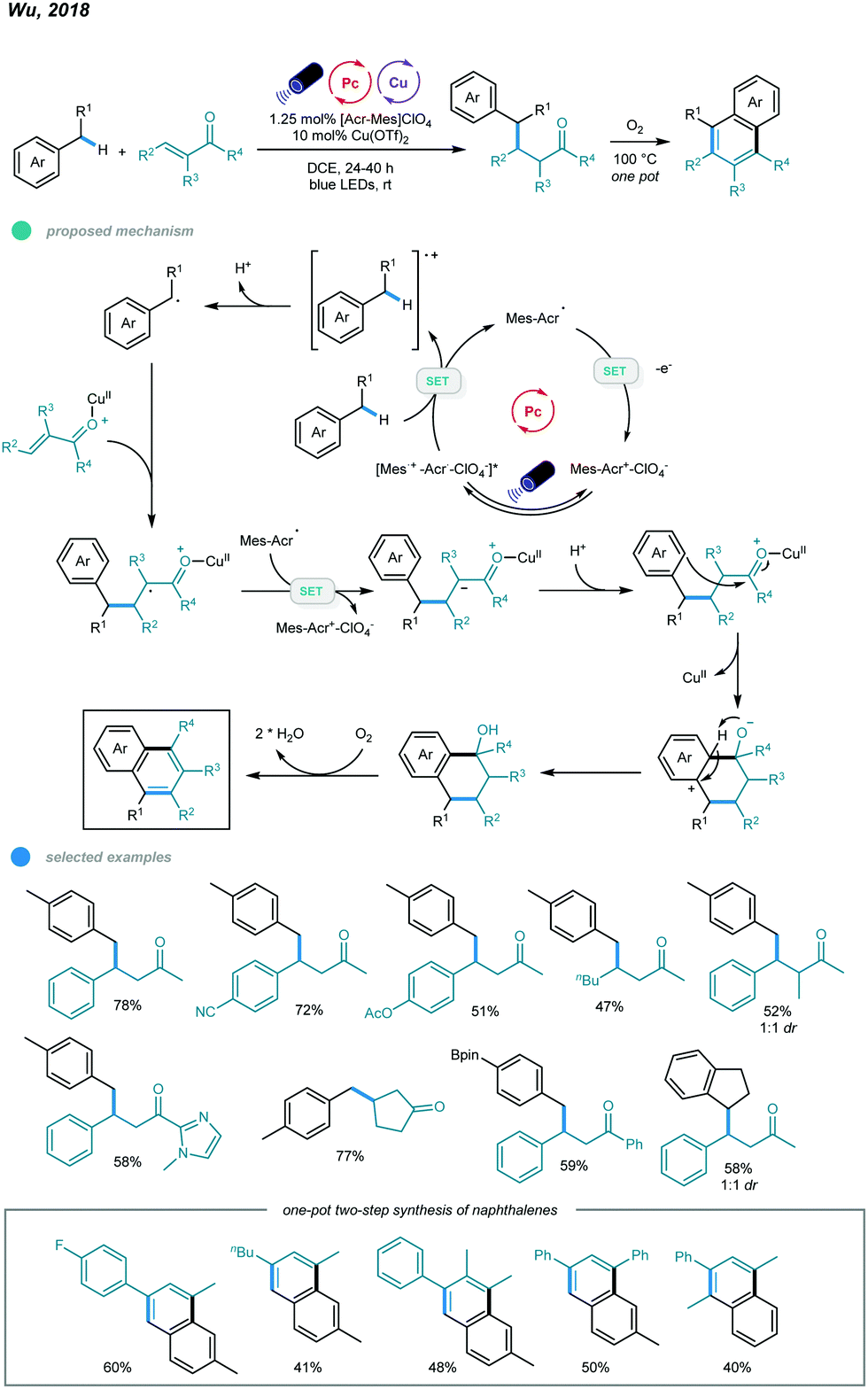 | ||
| Scheme 15 Photoredox and copper dual-catalyzed Giese reactions between benzylic C(sp3)–H and enones. | ||
More recently, asymmetric functionalization of C–H bonds in benzylic and allylic hydrocarbons, as well as in unactivated alkanes, was realized by Gong and coworkers (Scheme 16).54 The combination of a commercially available HAT organophotocatalyst (5,7,12,14-pentacenetetrone), earth-abundant metal (Cu or Co), tunable chiral bisoxazoline ligand and blue light led to transformations occurring with good efficiency and high regio- and stereoselectivity.
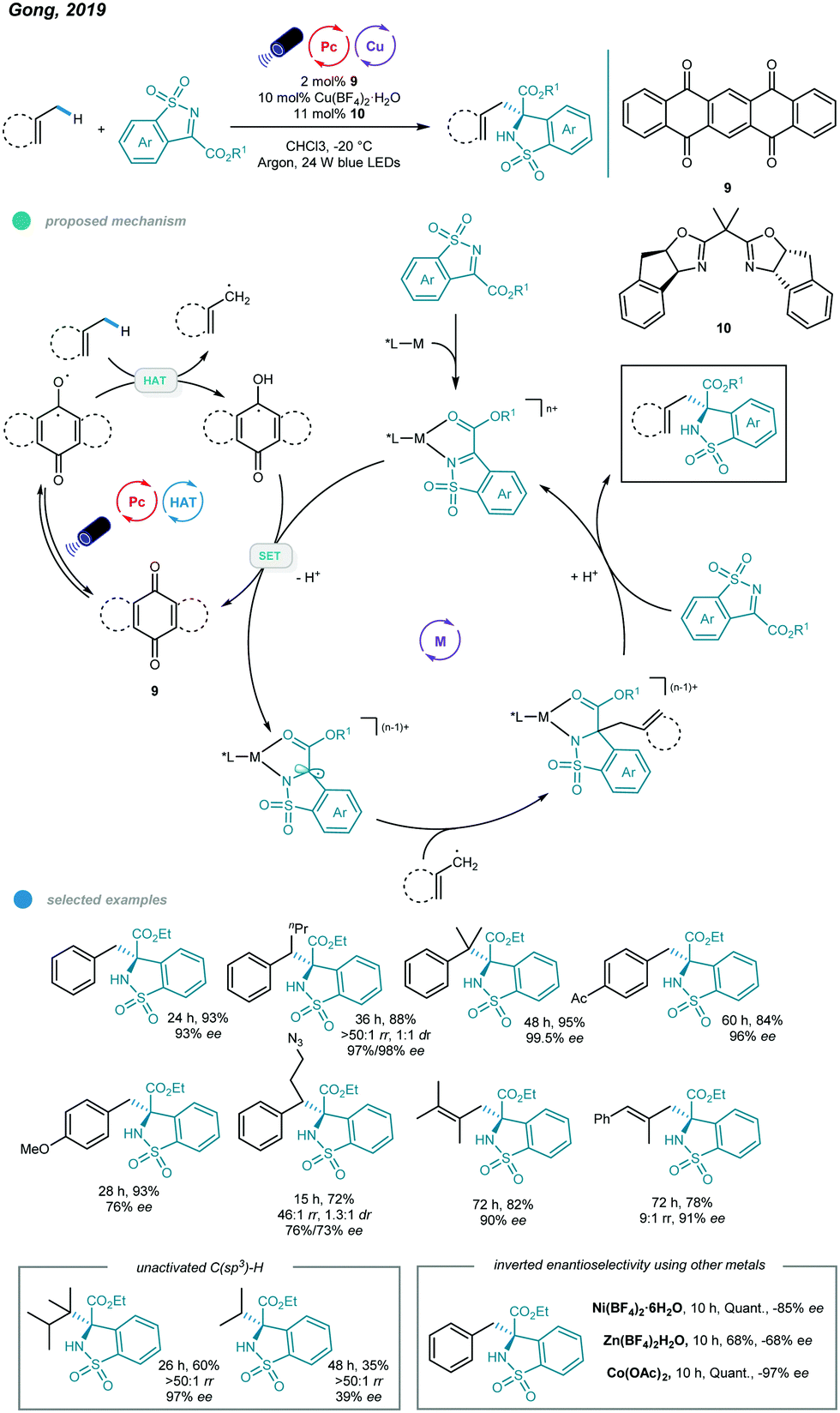 | ||
| Scheme 16 Photoredox and copper dual-catalyzed asymmetric C–H functionalizations of hydrocarbons with N-sulfonylimines. | ||
A wide range of functionalized chiral products was obtained economically by reacting N-sulfonylimines and hydrocarbons at −20 °C. The alkyl radical is generated along with a semiquinone-type radical via HAT between hydrocarbons and the excited organophotocatalyst. Then, the imine-coordinated chiral metal catalyst is reduced by the semiquinone-type radical to form a metal-stabilized carbon radical, which reacts with the alkyl radical to afford a new metal intermediate. At this junction, the regio- and enantioselectivity are sterically controlled by the chiral ligand-transition metal complex. Finally, the chiral product is delivered after protonation and ligand substitution, and the imine-coordinated chiral metal catalyst is regenerated. Primary, secondary, and tertiary benzylic hydrocarbons all worked well in this reaction, affording the corresponding chiral products in 76–97% yields with high enantioselectivities. However, secondary benzylic hydrocarbons showed no diastereoselectivity. Toluene-type substrates bearing halogen, ester, boron ester, ketone, and amide groups, as well as diversely substituted alkylbenzenes, all produced the desired products and exhibited excellent reactivities and enantioselectivities.
Moreover, a number of tri- and tetrasubstituted alkenes also underwent this asymmetric C–H functionalization and showed good yields with moderate to high ee values, albeit with a higher catalyst loading and reaction temperature and a long reaction time (3–5 days). Notably, the application of other nonprecious transition metals, including zinc, nickel, and cobalt, led to inverted enantioselectivities compared to those of a copper complex used with the same chiral ligand and identical reaction conditions, providing the possibility for development of stereodivergent syntheses by employing different metals. Moreover, late-stage modification of medicinal agents, including Celebrex, a Lopid derivative, and Skelaxin, afforded chiral products with good activity and enantioselectivity.
In 2018, an UV light induced benzylic C–H trifluoromethylation has been developed by Cook and coworkers using Grushin's reagent in an acetone/water solvent system (Scheme 17).55 UV light irradiation results in the generation of an active (bpy)Cu(CF3)2via hemolysis of the Grushin's reagent and sulfate radical anion from ammonium persulfate. The HAT event between the sulfate radical anion and benzylic C–H bond gives a benzylic radical. The resulting benzylic radical is intercepted by the active (bpy)Cu(CF3)2 to form a Bn–(bpy)Cu(CF3)2 complex, which undergoes subsequent reductive elimination to furnish the benzylic C–H trifluoromethylation product. Both primary and secondary benzylic C–H bonds could undergo this trifluoromethylation reaction smoothly. Typically, primary benzylic C–H bonds have a faster reaction rate and give the corresponding products in higher yields. Notably, late-stage C–H trifluoromethylation of bioactive molecules have also been successfully performed. However, tertiary benzylic C–H bonds showed no reactivity. Later on, using Grushin's reagent, the Hong group realized the direct C(sp3)–H trifluoromethylation of unactivated alkanes with the aid of an oxone as additive under the irradiation of blue LED, in which o examples of benzylic C(sp3)–H trifluoromethylation have been included (Scheme 17).56
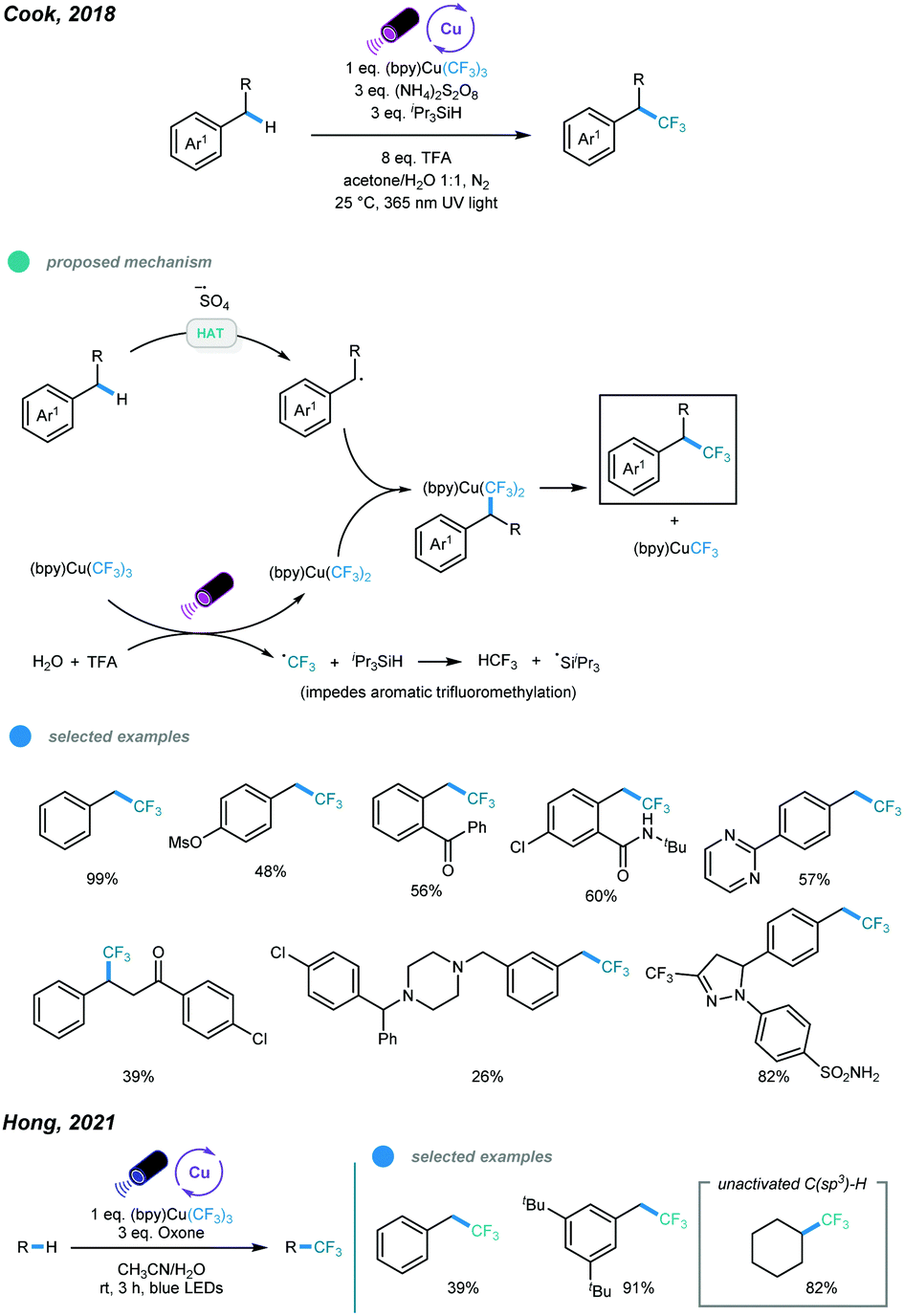 | ||
| Scheme 17 C(sp3)–H trifluoromethylation by trifluoromethyl copper complexes. | ||
Very recently, methylation of benzylic and α-amino C(sp3)–H bonds via photo/nickel dual catalysis has been reported by Stahl and coworkers (Scheme 18).57 Mechanistically, visible light-initiated triplet energy transfer between photocatalyst and di-tert-butyl peroxide (DTBP) leads to the generation of alkoxyl radicals, which could undergo either β-methyl scission to generate a methyl radical, or hydrogen-atom transfer (HAT) with the C(sp3)–H bond to generate a benzylic or α-amino radical. At last, the methylation product is formed via the cross-coupling of the resulting two types of radicals with the aid of nickel catalyst. A series of activated C–H substrates as well as α-amino substrates including medicinally relevant building blocks and drug molecules all could be methylated smoothly.
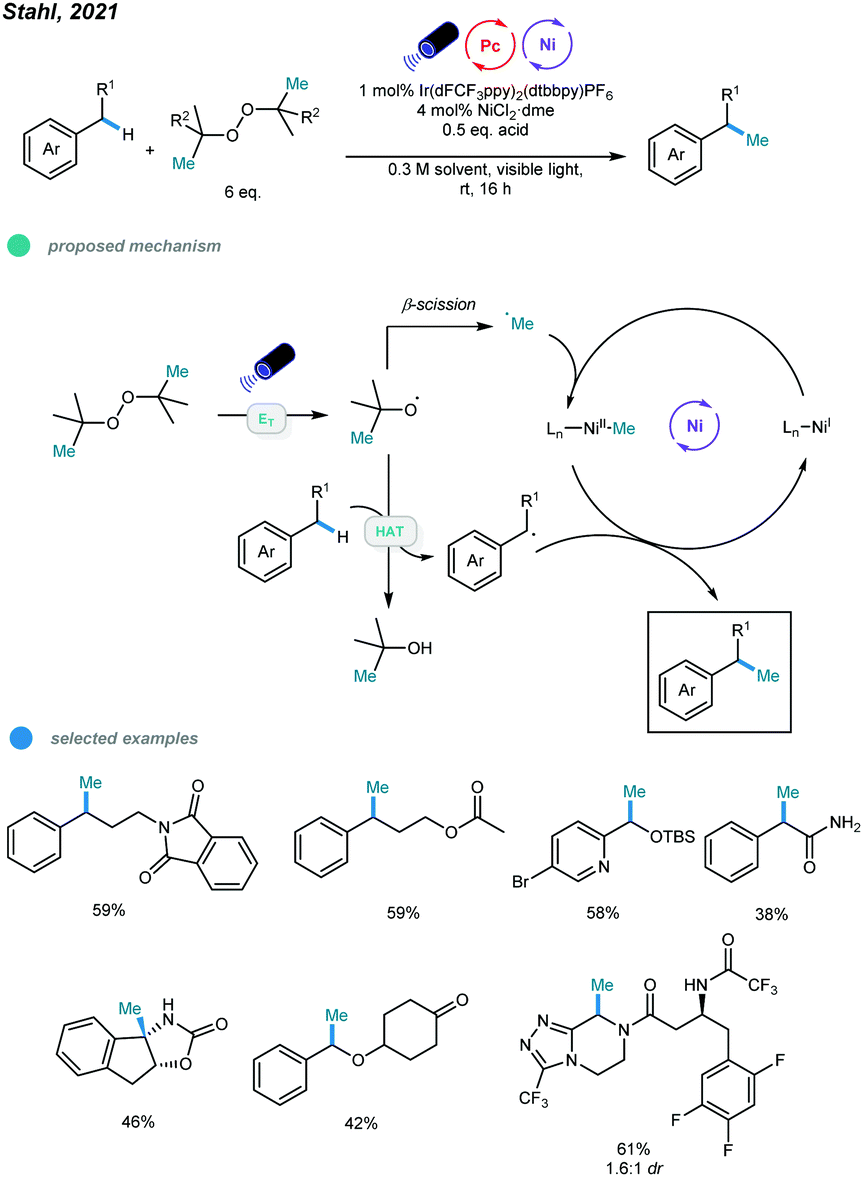 | ||
| Scheme 18 Photo and nickel dual-catalyzed benzylic and α-amino C(sp3)–H bonds methylation reactions. | ||
5. Carboxylation of benzylic and allylic C(sp3)–H bonds
The fixation of CO2 into organic skeletons via C–C bond formation has always been an appealing approach. In particular, directly fixing CO2 into readily available hydrocarbons remains more intriguing. In 2016, Murakami and coworkers used a combination of ketone photosensitizer and copper catalyst and realized a photoinduced carboxylation reaction between allylic C–H bonds of simple alkenes and CO2 (Scheme 19).58 The reaction was conducted at 110 °C under UV irradiation and with KtOBu as the base. The catalytic cycle starts with generation of an excited ketone via UV irradiation of the ketone photosensitizer. Abstraction of an allylic hydrogen from an alkene gives a geminate radical pair that is prone to undergo radical–radical coupling to form the tertiary homoallylic alcohol. The homoallylic alcohol is then deprotonated by copper tert-butoxide to form a copper alkoxide intermediate, and then β-carbon elimination furnishes an allyl copper species. Nucleophilic addition of the resulting allyl copper species to CO2 affords a copper carboxylate that undergoes ligand exchange with KtOBu to deliver the carboxylate salt and regenerate active copper tert-butoxide. Several cyclic and linear alkenes were used with moderate to good efficiency. It is worth noting that branched carboxylic acid isomers were favored products. | ||
| Scheme 19 Photoredox and copper/nickel dual-catalyzed benzylic and allylic C(sp3)–H carboxylation. | ||
However, when toluene derivatives containing benzylic C–H bonds were employed, no carboxylation product was observed. In 2019, the Murakami group extended the scope to benzylic C–H bonds by changing the metal from copper to nickel (Scheme 19).59 The combination of xanthone photosensitizer, NiCl2·6H2O metal catalyst, di(2-pyridyl)methane ligand, KtOBu base, benzene solvent, and irradiation with UV LED lamps (λmax = 365 nm) enabled carboxylation of toluene derivatives under one atmosphere of CO2 at room temperature. Herein, the excited xanthone abstracts a hydrogen atom from the benzylic C−H bond to give a benzylic radical that adds to the active Ni0 catalyst to afford a benzyl-NiI species. The resulting NiI species traps one molecule of carbon dioxide to give a NiI carboxylate, and then SET with a ketyl radical anion furnishes the carboxylate anion and regeneration of the active Ni0 species and the ground state ketone. A ring opening experiment with cyclopropylmethylanisole was carried out, and the acyclic carboxylic acid was the major product, indicating the involvement of benzylic radicals and thus supporting the proposed mechanism. A range of methylbenzene derivatives was tested, and monocarboxylated products were generated exclusively for all substrates. ortho-Methyl groups and halides, including fluoride and chloride, were all tolerated. However, the carboxylation reaction of ethylbenzene showed low efficiency, giving carboxylic acid in only 15% yield; this requires further improvement.
6. Alkynylation of allylic C(sp3)–H bonds
1,4-Enynes constitute a class of useful synthons that is important in organic synthesis. In 2020, Mejía and coworkers reported photoinduced copper-catalyzed cross-dehydrogenative coupling (CDC) for allylic C–H alkynylation (Scheme 20).60 A series of valuable 1,4-enynes was prepared via coupling of cyclic alkenes and aryl alkynes in a one-step process involving 5 mol% of a copper(I) terpyridyl complex as the photocatalyst, two equivalents of DTBP (di-tert-butylperoxide) as the oxidant and irradiation with white light (xenon lamp, λ = 300–700 nm) at room temperature. In the mechanism, [LCuI]+ is first converted to excited [LCuI]+* upon irradiation with white light. The excited copper species then undergoes SET with DTBP, producing an oxidized [LCuIIOR′]+ complex and R′O˙ radical. The R′O˙ radical abstracts a hydrogen atom from the C–H bond of the cyclic alkene, generating an allylic radical that is trapped by the oxidized [LCuIIOR′]+ complex to afford a CuIII intermediate. The CuIII intermediate undergoes ligand exchange with the organocuprate LCu-alkyne to produce a new CuIII complex that undergoes reductive elimination and delivers the CDC product with regeneration of the active [LCuI]+ catalyst. Alkynes bearing ortho-, active- and heterocyclic substituents all worked well in this system. Cyclic alkenes with different ring sizes were alkynylated smoothly. However, the C–H bonds of internal alkenes and aliphatic alkynes were not feasible substrates for this allylic C–H alkynylation protocol.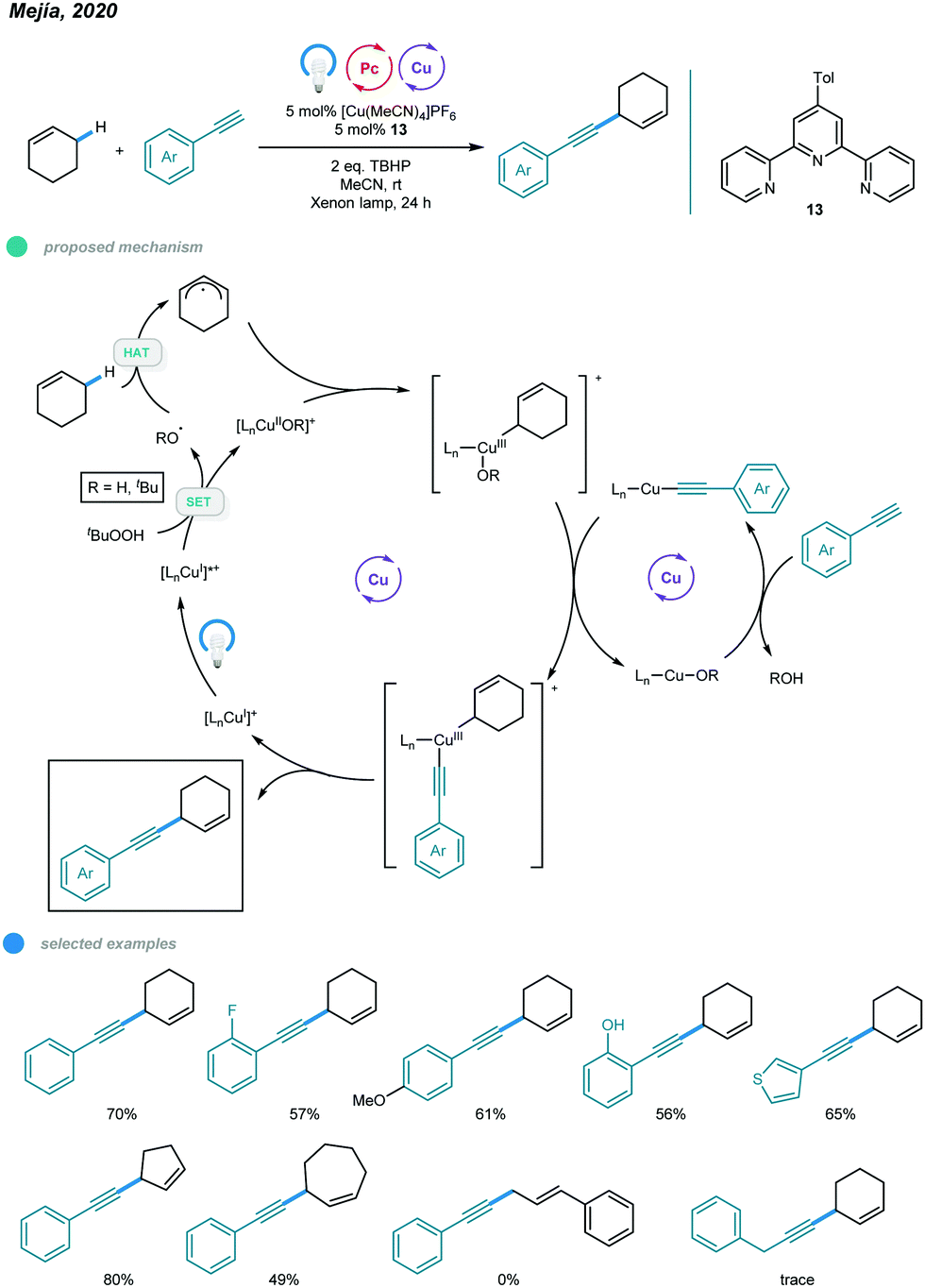 | ||
| Scheme 20 Photoredox and copper dual-catalyzed allylic C–H alkynylations. | ||
7. Sulfonylation and azidation of benzylic C(sp3)–H bonds
Although a series of two-component transformations giving C–C bond formation based on C(sp3)–H activation have been established via metallaphotocatalysis, protocols for C-heteroatom bond formation and three-component reactions remain undeveloped. Very recently, Gong and coworkers reported a photoredox and nickel dual-catalyzed three-component reaction involving a C(sp3)–H precursor, a SO2 surrogate and a common α,β-unsaturated carbonyl compound (Scheme 21).61 The application of a well-tailored chiral bisoxazoline ligand enabled the asymmetric three-component sulfonylation reaction, and a series of α-C chiral sulfones was prepared. (DABCO·(SO2)2) was found to be an efficient SO2 surrogate in this cascade protocol.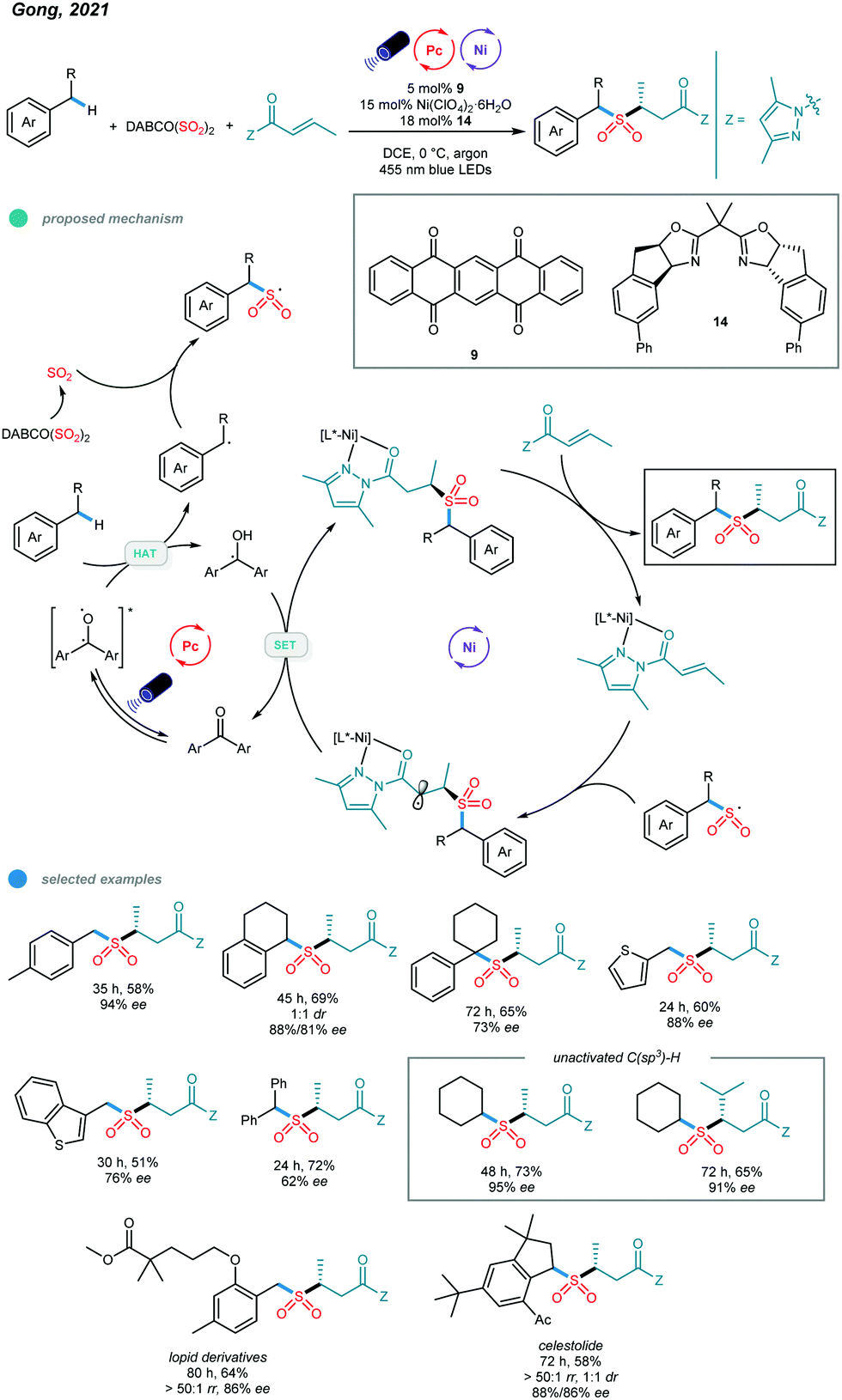 | ||
| Scheme 21 Photoredox and nickel dual-catalyzed three-component sulfonylations of C–H bonds of alkylbenzenes. | ||
Mechanistically, the chiral nickel catalyst L*-NiII first coordinates with the α,β-unsaturated N-acylpyrazole to form a new NiII complex. Additionally, the transient carbon radical generated from HAT with the excited organophotocatalyst is trapped by the sulfur dioxide released in situ to give the stable sulfonyl radical. The addition of the sulfonyl radical to the metal-coordinated Michael acceptor produces a metal-coordinated radical species, which undergoes a SET reduction with the semiquinone-type radical intermediate, and subsequent protonation affords the chiral sulfonylation product. In addition to alkanes, alkylbenzenes bearing primary, secondary, tertiary and benzylic C(sp3)–H bonds as well as methyl-substituted heterocycles all worked well in this asymmetric cascade reaction, providing the desired products in good yields with moderate to high enantioselectivities. Significantly, late-stage modifications of a Lopid derivative and Celestolide were accomplished with good yields and ee values.
Notably, by merging metal catalysis, electrocatalysis, and photocatalysis, Lei and coworkers realized manganese-catalyzed oxidative azidation of C(sp3)–H bonds (Scheme 22).62 This electrophotocatalytic protocol supports installation of synthetically useful azide groups into alkylbenzenes and simple alkanes while using nucleophilic NaN3 as an azide source under mild conditions. In this protocol, the benzylic radical was produced via HAT with either an excited organophotocatalyst or azide radical generated from anodic oxidation of NaN3. For the Mn catalytic cycle, initial ligand exchange of N3− with MnII(L) gives an N3-MnII(L) intermediate, which undergoes anodic oxidation to form an N3-MnIII(L) intermediate. Subsequent azide transfer from the N3-MnIII(L) intermediate to a benzylic radical delivers the azidation product. An array of tertiary and secondary benzyl C(sp3)–H was applied, and moderate to excellent yields were obtained. Heterocycles bearing secondary benzyl C(sp3)–H bonds, indane, and long-chain alkyl-substituted benzenes were all effective candidates. Notably, a scale-up reaction run on a 10 mmol scale proceeded smoothly and with good yield. This electrophotocatalytic protocol opens a new avenue allowing chemists to address some challenging organic syntheses that are difficult to realize with traditional methodologies.
 | ||
| Scheme 22 Manganese catalyzed oxidative azidation of C(sp3)–H bonds via electrophotocatalysis. | ||
8. Conclusion and outlook
In this feature article, we have highlighted the recent progress seen with metallaphoto-catalyzed functionalization of benzylic and allylic C(sp3)–H bonds. The newly developed approaches have been classified according to the type of transformation, including arylation, acylation, alkylation, carboxylation, alkynylation, sulfonylation and azidation. The illustrated methodologies enable the application of abundant hydrocarbons such as ethylbenzenes and alkenes as coupling partners in transition-metal catalyzed reactions. The transformations of more native functionalities to high-value compounds and the advantages of step and atom economy made these protocols appealing. The mild benzylic and allylic radical generating pathways involving HAT or SET allow the reactions to occur under very mild conditions, proceed with good chemo- and regioselectivity and illustrate good functional group compatibility. In some cases, an excess of C–H substrate is required for the transformations, possibly to boost the rate of bimolecular HAT and compete with radical annihilations. Future studies should focus on a few key challenges. First, the mechanism should be investigated in more detail since different research groups have described slightly different mechanisms for similar transformations. Second, compared to C–C bond formation, C-heteroatom bond formation via benzylic and allylic C(sp3)–H activation remains undeveloped and deserves more attention. Additionally, from the standpoint of practicability, more heterogeneous catalytic modes employing reusable photocatalysts should be developed. Moreover, considering the exclusive advantages of electrocatalysis, a combination of electro- or photoelectrocatalysis with C–H bond activation would be attractive. In addition, more effort should be dedicated to developing other efficient chiral ligands for asymmetric C–H bond functionalization and using benzylic and allylic radicals in more multicomponent reactions.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the King Abdullah University of Science and Technology (KAUST), Saudi Arabia, Office of Sponsored Research (URF/1/4384).Notes and references
- T. Rogge, N. Kaplaneris, N. Chatani, J. Kim, S. Chang, B. Punji, L. L. Schafer, D. G. Musaev, J. Wencel-Delord, C. A. Roberts, R. Sarpong, Z. E. Wilson, M. A. Brimble, M. J. Johansson and L. Ackermann, Nat. Rev. Methods Primers, 2021, 1, 43 CrossRef.
- P. Gandeepan, T. Müller, D. Zell, G. Cera, S. Warratz and L. Ackermann, Chem. Rev., 2018, 119, 2192–2452 CrossRef PubMed.
- L. Ackermann, Chem. Rev., 2011, 111, 1315–1345 CrossRef CAS PubMed.
- C. Sambiagio, D. Schönbauer, R. Blieck, T. Dao-Huy, G. Pototschnig, P. Schaaf, T. Wiesinger, M. F. Zia, J. Wencel-Delord and T. Besset, Chem. Soc. Rev., 2018, 47, 6603–6743 RSC.
- T. Gensch, M. Hopkinson, F. Glorius and J. Wencel-Delord, Chem. Soc. Rev., 2016, 45, 2900–2936 RSC.
- G. Rouquet and N. Chatani, Angew. Chem., Int. Ed., 2013, 52, 11726–11743 CrossRef CAS.
- J. Yamaguchi, A. D. Yamaguchi and K. Itami, Angew. Chem., Int. Ed., 2012, 51, 8960–9009 CrossRef CAS.
- R. Giri, B.-F. Shi, K. M. Engle, N. Maugel and J.-Q. Yu, Chem. Soc. Rev., 2009, 38, 3242–3272 RSC.
- X. Chen, K. M. Engle, D. H. Wang and J. Q. Yu, Angew. Chem., Int. Ed., 2009, 48, 5094–5115 CrossRef CAS PubMed.
- J. He, M. Wasa, K. S. Chan, Q. Shao and J.-Q. Yu, Chem. Rev., 2017, 117, 8754–8786 CrossRef CAS PubMed.
- R. R. Karimov and J. F. Hartwig, Angew. Chem., Int. Ed., 2018, 57, 4234–4241 CrossRef CAS PubMed.
- K. Liao, S. Negretti, D. G. Musaev, J. Bacsa and H. M. Davies, Nature, 2016, 533, 230–234 CrossRef CAS PubMed.
- C. Le, Y. Liang, R. W. Evans, X. Li and D. W. MacMillan, Nature, 2017, 547, 79–83 CrossRef CAS.
- F.-L. Zhang, K. Hong, T.-J. Li, H. Park and J.-Q. Yu, Science, 2016, 351, 252–256 CrossRef CAS.
- J. He, S. Li, Y. Deng, H. Fu, B. N. Laforteza, J. E. Spangler, A. Homs and J.-Q. Yu, Science, 2014, 343, 1216–1220 CrossRef CAS PubMed.
- H. Yi, G. Zhang, H. Wang, Z. Huang, J. Wang, A. K. Singh and A. Lei, Chem. Rev., 2017, 117, 9016–9085 CrossRef CAS PubMed.
- A. Hu, J.-J. Guo, H. Pan and Z. Zuo, Science, 2018, 361, 668–672 CrossRef CAS.
- J. M. Narayanam and C. R. Stephenson, Chem. Soc. Rev., 2011, 40, 102–113 RSC.
- D. Ravelli, S. Protti and M. Fagnoni, Chem. Rev., 2016, 116, 9850–9913 CrossRef CAS.
- J. Xuan and W. J. Xiao, Angew. Chem., Int. Ed., 2012, 51, 6828–6838 CrossRef CAS.
- C. Zhu, H. Yue, L. Chu and M. Rueping, Chem. Sci., 2020, 11, 4051–4064 RSC.
- C. Zhu, H. Yue, J. Jia and M. Rueping, Angew. Chem., Int. Ed., 2021, 60, 17810–17831 CrossRef CAS PubMed.
- C. K. Prier, D. A. Rankic and D. W. MacMillan, Chem. Rev., 2013, 113, 5322–5363 CrossRef CAS PubMed.
- J. Twilton, P. Zhang, M. H. Shaw, R. W. Evans and D. W. MacMillan, Nat. Rev. Chem., 2017, 1, 1–19 CrossRef.
- J. C. Tellis, C. B. Kelly, D. N. Primer, M. Jouffroy, N. R. Patel and G. A. Molander, Acc. Chem. Res., 2016, 49, 1429–1439 CrossRef CAS.
- N. Holmberg-Douglas and D. A. Nicewicz, Chem. Rev., 2021 DOI:10.1021/acs.chemrev.1021c00311.
- L. Capaldo, D. Ravelli and M. Fagnoni, Chem. Rev., 2021 DOI:10.1021/acs.chemrev.1021c00263.
- H. Cao, X. Tang, H. Tang, Y. Yuan and J. Wu, Chem. Catal., 2021, 1, 523–598 CrossRef.
- M.-M. Zhang, Y.-N. Wang, L.-Q. Lu and W.-J. Xiao, Trends Chem., 2020, 2, 764–775 CrossRef CAS.
- L. Mantry, R. Maayuri, V. Kumar and P. Gandeepan, Beilstein J. Org. Chem., 2021, 17, 2209–2259 CrossRef CAS.
- L. Guillemard and J. Wencel-Delord, Beilstein J. Org. Chem., 2020, 16, 1754–1804 CrossRef CAS.
- M. H. Shaw, V. W. Shurtleff, J. A. Terrett, J. D. Cuthbertson and D. W. MacMillan, Science, 2016, 352, 1304–1308 CrossRef CAS PubMed.
- D. R. Heitz, J. C. Tellis and G. A. Molander, J. Am. Chem. Soc., 2016, 138, 12715–12718 CrossRef CAS PubMed.
- B. J. Shields and A. G. Doyle, J. Am. Chem. Soc., 2016, 138, 12719–12722 CrossRef CAS.
- N. Ishida, Y. Masuda, N. Ishikawa and M. Murakami, Asian J. Org. Chem., 2017, 6, 669–672 CrossRef CAS.
- L. Huang and M. Rueping, Angew. Chem., Int. Ed., 2018, 57, 10333–10337 CrossRef CAS.
- A. Dewanji, P. E. Krach and M. Rueping, Angew. Chem., Int. Ed., 2019, 58, 3566–3570 CrossRef CAS PubMed.
- I. B. Perry, T. F. Brewer, P. J. Sarver, D. M. Schultz, D. A. DiRocco and D. W. MacMillan, Nature, 2018, 560, 70–75 CrossRef CAS.
- Y. Shen, Y. Gu and R. Martin, J. Am. Chem. Soc., 2018, 140, 12200–12209 CrossRef.
- W. Kim, J. Koo and H. G. Lee, Chem. Sci., 2021, 12, 4119–4125 RSC.
- X. Cheng, H. Lu and Z. Lu, Nat. Commun., 2019, 10, 3549 CrossRef.
- X. Cheng, T. Li, Y. Liu and Z. Lu, ACS Catal., 2021, 11, 11059–11065 CrossRef CAS.
- L. K. Ackerman, J. I. Martinez Alvarado and A. G. Doyle, J. Am. Chem. Soc., 2018, 140, 14059–14063 CrossRef CAS PubMed.
- P. E. Krach, A. Dewanji, T. Yuan and M. Rueping, Chem. Commun., 2020, 56, 6082–6085 RSC.
- T. Kawasaki, N. Ishida and M. Murakami, J. Am. Chem. Soc., 2020, 142, 3366–3370 CrossRef CAS.
- L. Huan, X. Shu, W. Zu, D. Zhong and H. Huo, Nat. Commun., 2021, 12, 3536 CrossRef CAS PubMed.
- Y. Okude, S. Hirano, T. Hiyama and H. Nozaki, J. Am. Chem. Soc., 1977, 99, 3179–3181 CrossRef CAS.
- K. Namba, J. Wang, S. Cui and Y. Kishi, Org. Lett., 2005, 7, 5421–5424 CrossRef CAS.
- A. Fürstner and N. Shi, J. Am. Chem. Soc., 1996, 118, 12349–12357 CrossRef.
- J. L. Schwarz, F. Schäfers, A. Tlahuext-Aca, L. Lückemeier and F. Glorius, J. Am. Chem. Soc., 2018, 140, 12705–12709 CrossRef CAS.
- H. Mitsunuma, S. Tanabe, H. Fuse, K. Ohkubo and M. Kanai, Chem. Sci., 2019, 10, 3459–3465 RSC.
- S. Tanabe, H. Mitsunuma and M. Kanai, J. Am. Chem. Soc., 2020, 142, 12374–12381 CrossRef CAS.
- H. Liu, L. Ma, R. Zhou, X. Chen, W. Fang and J. Wu, ACS Catal., 2018, 8, 6224–6229 CrossRef CAS.
- Y. Li, M. Lei and L. Gong, Nat. Catal., 2019, 2, 1016–1026 CrossRef CAS.
- S. Guo, D. I. AbuSalim and S. P. Cook, J. Am. Chem. Soc., 2018, 140, 12378–12382 CrossRef CAS.
- G. Choi, G. S. Lee, B. Park, D. Kim and S. H. Hong, Angew. Chem., 2021, 133, 5527–5534 CrossRef.
- A. Vasilopoulos, S. W. Krska and S. S. Stahl, Science, 2021, 372, 398–403 CrossRef CAS PubMed.
- N. Ishida, Y. Masuda, S. Uemoto and M. Murakami, Chem. – Eur. J., 2016, 22, 6524–6527 CrossRef CAS.
- N. Ishida, Y. Masuda, Y. Imamura, K. Yamazaki and M. Murakami, J. Am. Chem. Soc., 2019, 141, 19611–19615 CrossRef CAS.
- A. A. Almasalma and E. Mejía, Synthesis, 2020, 529–536 CAS.
- S. Cao, W. Hong, Z. Ye and L. Gong, Nat. Commun., 2021, 12, 2377 CrossRef CAS PubMed.
- L. Niu, C. Jiang, Y. Liang, D. Liu, F. Bu, R. Shi, H. Chen, A. D. Chowdhury and A. Lei, J. Am. Chem. Soc., 2020, 142, 17693–17702 CrossRef CAS PubMed.
Footnote |
| † These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2022 |