
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Resolving alternative structure determinations of indapamide using 13C solid-state NMR†‡
Caitlin L.
Evans
 ,
Ivana Radosavlijević
Evans
and
Paul
Hodgkinson
*
,
Ivana Radosavlijević
Evans
and
Paul
Hodgkinson
*
Department of Chemistry, Durham University, Stockton Road, Durham, DH1 3LE, UK. E-mail: paul.hodgkinson@durham.ac.uk
First published on 28th March 2022
Abstract
The conflict between alternative crystal structures in the Cambridge Structural Database for the diuretic drug indapamide hemihydrate (IND) has been resolved with the aid of 13C solid-state NMR. IND is seen to contain multiple distinct molecules in the asymmetric unit (Z′ = 4) rather than exhibiting disorder in the orientation of sulfonamide groups. The NMR crystallographic approach is a more effective tool for distinguishing between alternative structures than naïve judgements of quality based on crystallographic refinement agreement factors.
Repositories of crystal structure data, such as the Cambridge Structural Database (CSD),1 are invaluable resources for researchers working in diverse areas of molecular solid-state chemistry and materials science. As discussed in a recent survey2 of over 3000 “repeat structures” in the CSD, it is increasingly common to find multiple structure determinations for what appears to be the same solid form. This creates an obvious dilemma for an ordinary user of structural databases. The problem is compounded when there are questions of disorder in the crystal structure. Disorder is not uncommon, with about 6% of structures published in CSD between 2000 and 2017 containing comments (added during data curation) about disorder. While some of this disorder may be crystallographically and chemically uninteresting (e.g., alternative positioning of hydrogen atoms in methyl groups), other types of disorder are structurally significant.
A particularly interesting subset of the CSD disorder comments, found in about 10% of cases, refer to “disorder by symmetry”. This is used for cases when a fragment of the structure is disordered across two (or more) sets of atomic positions that do not individually reflect the space group symmetry; only the superposition of possible positions respects the symmetry. Such “disorder by symmetry” either means that the choice of space group or unit cell is incorrect (a crystallographic error), or that this fragment is indeed disordered. While additional diffraction experiments, including at different temperatures, can help identify the presence of disorder, due to dynamics or the incorrect selection of space group or unit cell, the user of an individual crystallographic result cannot resolve this ambiguity. In this context, spectroscopic techniques, which probe local chemical environments, can resolve the nature of crystallographic disorder, without requiring new diffraction-quality single crystals.3–5
Indapamide is a classic diuretic drug used in the treatment of hypertension,6,7 and is marketed as a crystalline hemihydrate, hereafter IND. Previous studies have focussed on the solid-state characterisation of additional forms of IND,8–10 while recent work has considered the amorphous11–14 and cocrystal forms.14,15 Reflecting the importance of solid-state characterisation for drug approval, there are multiple structures for IND in the CSD. We show here how 13C solid-state NMR readily identifies the most plausible crystal structure and confirm this with a new diffraction study.
There are three deposited crystal structures for IND in the CSD (Version 5.40), each with a distinct reference code. The oldest structure, FOCCAD,9 was derived from powder X-ray diffraction (PXRD) data. The VAGKUM10 structure was the result of a detailed single crystal structure determination (SCXRD) of IND, whilst most recently, WOCPEM14 was derived serendipitously from an unsuccessful attempt to co-crystallise IND with gliclazide. Table 1 summarises the key crystallographic data.
| FOCCAD | VAGKUM | WOCPEM | WOCPEM NEW | |
|---|---|---|---|---|
| a Space group in the CSD. Original reference gives I2/a. | ||||
| Formula unit | C16H16ClN3O3S·0.35H2O | C16H16ClN3O3S·½H2O | ||
| M r / g mol−1 | 372 | 374.83 | ||
| Temperature / K | 298 | 100 | 150 | 120 |
| Wavelength / Å | 1.54 | 1.54 | 0.71 | 0.71 |
| Crystal system | Monoclinic | Monoclinic | Monoclinic | Monoclinic |
| Space group | P2/aa | I2/a | P21/c | P21/c |
| a / Å | 23.811(3) | 15.059(9) | 30.060(11) | 30.140(10) |
| b / Å | 9.6940(9) | 9.6218(6) | 9.6685(3) | 9.6025(4) |
| c / Å | 15.114(2) | 23.508(14) | 23.573(10) | 23.461(8) |
| β / ° | 91.66(3) | 92.60(16) | 92.33(4) | 92.59(10) |
| V / Å3 | 3487.2(3) | 3402.6(4) | 6845.4(5) | 6783.2(4) |
| Z, Z′ | 4, 1 | 8, 1 | 16, 4 | 16, 4 |
| Data/restraints/parameters | (See text) | 3036 | 12492 | 18001 |
| 3 | 41 | 61 | ||
| 261 | 948 | 979 | ||
| R 1 [I ≥ 2σ(I)] | 0.0560 | 0.0369 | 0.0868 | 0.0842 |
There are well-known challenges in determining small-molecule crystal structures from PXRD data, but with careful data collection and analysis, robust structure determinations can give reliable structural models.16,17 Moreover, NMR is strongly complementary to PXRD, either for aiding structural determination or for validating derived structures.3 The FOCCAD structure determination omitted hydrogen atoms in the structure solution step, which involved direct space Monte Carlo simulated annealing with only 9 variable parameters (3 translational, 3 orientational and 3 torsion angles). In the structural refinement step, the hydrogen atoms were placed geometrically, leading to two additional refineable torsion angles, but there was no modelling of the sulfonamide group disorder. This approach gave an excellent Rietveld fit to the observed PXRD data,9 but DFT geometry optimisation of the determined structure resulted in significant atomic displacements (see Table S1, ESI† and associated discussion). Hence, FOCCAD was not considered further.
The VAGKUM and WOCPEM structures are closely related, and, as shown in Fig. S1 (ESI†), have virtually identical simulated PXRD patterns. VAGKUM has a unit cell volume that is a factor of two smaller, and involves disorder where the sulfonamide group is disordered over two positions with almost equal occupancies. The water molecule is also “disordered by symmetry” (see Fig. S3 and S4, ESI†). WOCPEM has a larger unit cell, with 4 formula units in the asymmetric unit (Z′ = 4), as opposed to Z′ = 1 for VAGKUM. This larger unit cell has no disorder, but the R factor is considerably larger (9% compared to 4%). Overlaying the four IND molecules in the WOCPEM asymmetric unit, Fig. 1 (b), shows that they adopt an identical conformation, differing only in the sulfonamide group orientation.
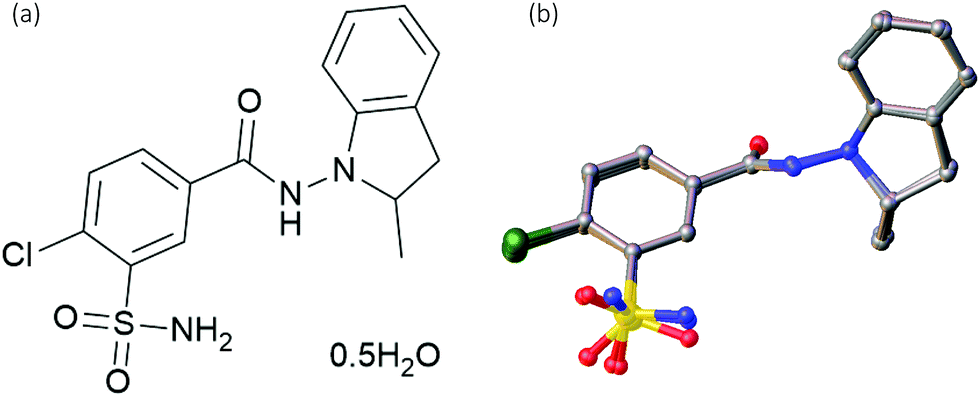 | ||
| Fig. 1 (a) Molecular structure of the formula unit of indapamide hemihydrate. (b) Overlay of the four distinct indapamide molecules in the asymmetric unit of WOCPEM (see text). | ||
VAGKUM and WOCPEM essentially describe the same structure but differ in how the sulfonamide group is modelled. This is an example of a molecular organic crystal exhibiting pseudo-symmetry.18–21 The four crystallographically independent molecules in the asymmetric unit are related by symmetry except for differences in the sulfonamide group orientation, which could quite plausibly be explained by disorder. The larger unit cell of WOCPEM also removes the “disorder by symmetry” of the water molecule, but this too could be the result of disorder rather than indicating a problem with the structure determination.
Without new evidence, either from repeat single-crystal studies or complementary experimental techniques, the choice of the most plausible structural model is not straightforward, especially as such two-fold disorder of the sulfonamide group is common.22–24 Disorder of included water molecules is also chemically plausible and consistent with the large ADPs (Fig. S3–S5, ESI†), so there is no argument to be made based on chemical intuition. The R factors are also not decisive; as discussed further below, a lower R factor does not necessarily indicate a more reliable structural model.25
NMR crystallography refers to the use of solid-state NMR, often in conjunction with first principles calculations, to resolve crystallographic questions.3,5 IND provides an example of the complementary role of diffraction and spectroscopic experiments. The presence of multiple resonances per carbon site in the 13C NMR spectrum, Fig. 2, clearly indicates that Z′ has to be greater than 1. The highlighted line shapes are most obviously explained by Z′ = 4. Although not strictly necessary to establish that Z′ > 1, calculated chemical shifts26 based on the WOCPEM structure show good agreement with the experimental data (as shown in Fig. S7, ESI†); the RMSD between experimental and calculated 13C chemical shifts of 1.70 ppm is in line with literature precedents.27,28 The calculations also helpfully confirm that the structure at ambient temperature (NMR data) is essentially indistinguishable from the structure at the measurement temperature for the single-crystal diffraction data (120–150 K).
 | ||
| Fig. 2 13C CP/MAS NMR spectrum of IND. Highlighted peaks clearly indicate Z′ > 1. Blue tick marks indicate DFT-calculated chemical shifts (using plane-wave basis sets) for the WOCPEM structure, whilst insets show deconvolution of the Me and CO signals into four components (see ESI† for further details). | ||
As would be expected from Fig. 1(b), the differences in chemical shift between corresponding carbon sites within the asymmetric unit are small, and so not all peaks show resolved “crystallographic splittings”. As previously observed, however, the magnitudes of the crystallographic splittings are predicted extremely well by the DFT calculation.29 It is worth noting the sizes of the systems that can now be handled using first principles calculation; WOCPEM contains 280 atoms in its unit cell. Further details of the computation, including modifications required to run calculations of this size can be found in the ESI.†
The NMR studies only require powered samples and so can be applied directly on the commercial material, without needing to obtain diffraction-quality single crystals. However, one of the crystallisation routes14 attempted in this work did provide diffraction-quality single crystals (see ESI†). The resulting structure, labelled here as WOCPEM NEW (CCDC deposition number: 2115849), was in excellent agreement with WOCPEM (Table 1).
The clear conclusion from the NMR data is that there is no disorder of the sulfonamide group, either static (which would lead to structure-less broadening of the 13C resonances) or dynamic (which would lead to sharpened single resonances).3 In terms of diffraction, the ordering of the sulfonamide orientation leads to a doubling of the unit cell and weak additional reflections. However, the typical superstructure reflection intensities, such as (300), (310) and (111), were 3 to 4 orders of magnitude weaker than other intensities for WOCPEM NEW, and these reflections must have been overlooked when determining the VAGKUM structure. Deposited reflection intensity data for the smaller unit cell would not allow this problem to be corrected, since the key reflections would normally be missing from the data set.
The fact that the WOCPEM structures, which are independently verified by repeat single-crystal X-ray diffraction and solid-state NMR experiments, have higher R factors (see Table 1) than the alternative VAGKUM structure determination may seem counter-intuitive; a non-expert user of crystallographic databases might assume that a lower R implies a “better” structure. There are multiple factors, however, that complicate comparison of the R factors. For example, the shorter wavelength used for the WOCPEM studies means these involve a much larger number of reflections (including many weak reflections associated with the pseudo-symmetry). Combined with the very different unit cell sizes, this means that the hkl data sets are not easily compared. Overall, the R factor for the WOCPEM structures is reasonable for standard single-crystal diffraction structure of an average, middle-size organic molecule. Relaxing the number of restraints (from 61 for WOCPEM NEW) could reduce the R value but produce a less chemically sensible solution. This highlights the risks of using R naïvely to assess the relative correctness of different structural solutions.
In summary, we have described how solid-state NMR can be straightforwardly used to distinguish between conflicting structures in structural databases. NMR crystallographic methods are particularly complementary to diffraction when resolving potential issues of “disorder by symmetry” and pseudo-symmetry, since these are often associated with differences in the number of molecules in the asymmetric unit.
This work was funded by EPSRC DTP (Grant number: EP/R513039/1) and benefited from the software tools developed by the Collaborative Computational Project for NMR Crystallography (Grant number: EP/M022501/1). We thank the Durham University Crystallography Service for acquisition and solution of WOCPEM NEW, Mr Gary Oswald for acquisition of PXRD data and Prof. John S. O. Evans for helpful discussions.
Conflicts of interest
The authors declare no conflicts of interest.References
- C. R. Groom, I. J. Bruno, M. P. Lightfoot and S. C. Ward, Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater., 2016, 72, 171–179 CrossRef CAS PubMed.
- C. M. Widdifield, J. D. Farrell, J. C. Cole, J. A. K. Howard and P. Hodgkinson, Chem. Sci., 2020, 11, 2987–2992 RSC.
- P. Hodgkinson, Prog. Nucl. Magn. Reson. Spectrosc., 2020, 118–119, 10–53 CrossRef CAS PubMed.
- R. F. Moran, D. M. Dawson and S. E. Ashbrook, Int. Rev. Phys. Chem., 2017, 36, 39–115 Search PubMed.
- D. L. Bryce, IUCrJ, 2017, 4, 350–359 Search PubMed.
- G. C. Roush, M. E. Ernst, J. B. Kostis, S. Tandon and D. A. Sica, Hypertension, 2015, 65, 1041–1046 CrossRef CAS PubMed.
- G. C. Roush and D. A. Sica, Am. J. Hypertens., 2016, 29, 1130–1137 CrossRef CAS PubMed.
- P. Ghugare, V. Dongre, P. Karmuse, R. Rana, D. Singh, A. Kumar and Z. Filmwala, J. Pharm. Biomed. Anal., 2010, 51, 532–540 CrossRef CAS PubMed.
- M. Smrkolj and A. Meden, Pharmazie, 2006, 61, 999–1004 CAS.
- J. Bojarska, A. Fruziński and W. Maniukiewicz, J. Mol. Struct., 2016, 1116, 22–29 CrossRef CAS.
- A. Drogoń, M. Skotnicki, A. Skotnicka and M. Pyda, Pharmaceutics, 2020, 12, 800–815 CrossRef PubMed.
- M. Skotnicki, A. Drogoń, J. J. Calvin, P. F. Rosen, B. F. Woodfield and M. Pyda, Thermochim. Acta, 2019, 674, 36–43 CrossRef CAS.
- Z. Wojnarowska, K. Grzybowska, L. Hawelek, M. Dulski, R. Wrzalik, I. Gruszka, M. Paluch, K. Pienkowska, W. Sawicki, P. Bujak, K. J. Paluch, L. Tajber and J. Markowski, Mol. Pharmaceutics, 2013, 10, 3612–3627 CrossRef CAS PubMed.
- M. Aljohani, P. MacFhionnghaile, P. McArdle and A. Erxleben, Int. J. Pharm., 2019, 561, 35–42 CrossRef CAS PubMed.
- S. Allu, K. Suresh, G. Bolla, M. K. C. Mannava and A. Nangia, CrystEngComm, 2019, 21, 2043–2048 RSC.
- K. D. M. Harris, Top. Curr. Chem., 2012, 315, 133–178 CrossRef CAS PubMed.
- R. Černý, Crystals, 2017, 7, 142–152 CrossRef.
- P. H. Zwart, R. W. Grosse-Kunstleve, A. A. Lebedev, G. N. Murshudov and P. D. Adams, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2008, 64, 99–107 CrossRef CAS PubMed.
- A. A. Lebedev and M. N. Isupov, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2014, 70, 2430–2443 CrossRef CAS PubMed.
- C. Giacovazzo, H. L. Monaco, G. Artioli, D. Viterbo, M. Milanesio, G. Ferraris, G. Gilli, P. Gilli, G. Zanotti and M. Catti, Fundamentals of Crystallography, 3rd edn, Oxford University Press, Oxford, UK, 2011 Search PubMed.
- J. Li and J. Sun, Acc. Chem. Res., 2017, 50, 2737–2745 CrossRef CAS PubMed.
- H. E. Kerr, L. K. Softley, K. Suresh, A. Nangia, P. Hodgkinson and I. R. Evans, CrystEngComm, 2015, 17, 6707–6715 RSC.
- B. I. Harriss, L. Vella-Zarb, C. Wilson and I. R. Evans, Cryst. Growth Des., 2014, 14, 783–791 CrossRef CAS.
- X. Filip, G. Borodi and C. Filip, Phys. Chem. Chem. Phys., 2011, 13, 17978–17986 RSC.
- C. Buchsbaum and M. U. Schmidt, Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater., 2007, 63, 926–932 CrossRef CAS PubMed.
- S. J. Clark, M. D. Segall, C. J. Pickard, P. J. Hasnip, M. I. J. Probert, K. Refson and M. C. Payne, Z. Kristallogr., 2005, 220, 567–570 CAS.
- J. D. Hartman, R. A. Kudla, G. M. Day, L. J. Mueller and G. J. O. Beran, Phys. Chem. Chem. Phys., 2016, 18, 21686–21709 RSC.
- E. Salager, G. M. Day, R. S. Stein, C. J. Pickard, B. Elena and L. Emsley, J. Am. Chem. Soc., 2010, 132, 2564–2566 CrossRef CAS PubMed.
- A. Abraham, D. C. Apperley, S. J. Byard, A. J. Ilott, A. J. Robbins, V. Zorin, R. K. Harris and P. Hodgkinson, CrystEngComm, 2016, 18, 1054–1063 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available: Includes materials, methods, experimental PXRD data, computational methods and visualisation of structural models. CCDC 2115849. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1cc06256e |
| ‡ A separate archive of research data for this article can be found at DOI: 10.15128/r2df65v796t |
| This journal is © The Royal Society of Chemistry 2022 |