
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
C,C′-Ru to C,B′-Ru isomerisation in bis(phosphine)Ru complexes of [1,1′-bis(ortho-carborane)]†‡
Rebekah J.
Jeans
 ,
Georgina M.
Rosair
and
Alan J.
Welch
*
,
Georgina M.
Rosair
and
Alan J.
Welch
*
Institute of Chemical Sciences, Heriot-Watt University, Edinburgh EH14 4AS, UK. E-mail: a.j.welch@hw.ac.uk
First published on 1st December 2021
Abstract
We report herein the first example of the controlled isomerisation of a C,C′-bound (to metal) bis(ortho-carborane) ligand to C,B′-bound with no other change in the molecule. Since the C and B vertices of carboranes have different electron-donating properties this transformation allows the reactivity of the metal centre to be fine-tuned.
Carboranes are exceedingly versatile ligands to transition-metals.1 Deboronation of the neutral carborane [closo-1,2-C2B10H12] to the anion [nido-7,8-C2B9H11]2− affords a ligand which is isolobal with the ubiquitous Cp−, able to bind to metals in full η5 fashion,2 or η<5 in slipped metallacarboranes.3 Alternatively carboranes, particularly anionic carboranes, are able to co-ordinate metals through one,4 two5 or three6 B–H⇀M B–agostic interactions, taking advantage of the hydridic nature of H atoms bonded to B. Finally direct C–M7 and B–M8 sigma bonding is well established and is sometimes accompanied by B–H⇀M B–agostic bonding from adjacent B atoms.9
Whether a carborane binds directly to a metal, or to a substituent which is subsequently linked to a metal, through a C or B vertex is particularly important in that, everything else being equal, a B-bound carborane is more electron-donating than a C-bound carborane.10 This affords two isomeric forms of the same ligand which are isosteric but not isoelectronic, and recently this has been exploited to fine-tune the properties of metal–carborane complexes.11
Bis(carboranes) are molecules composed of two carborane moieties connected by a direct C–C, C–B or B–B bond and, of the various possible bis(carboranes), [1-(1′-closo-1′,2′-C2B10H11)-closo-1,2-C2B10H11] or more simply [1,1′-bis(ortho-carborane)], is the most studied and has undergone a resurgence of interest in recent years.12 Following double deprotonation at the protonic C2H and C2′H sites, [1,1′-bis(ortho-carborane)] can be used as a κ2 chelating ligands in both homoleptic13 and heteroleptic9b,11c,d,12f,14 transition-metal complexes.
In 2016 we reported catalytically-active (arene)Ru complexes of doubly-deprotonated [1,1′-bis(ortho-carborane)] in which the metal coordination was completed by a B3′–H⇀Ru B–agostic interaction.9b Reaction of these compounds with phosphine (2 × PPh3 or dppe) resulted in displacement of the arene, coordination of the phosphine and a change in the ligating mode of the bis(carborane) from X2(C,C′)L (L = agostic interaction) to X2(C,B′)L, the first time C,B′ ligation of [1,1′-bis(ortho-carborane)] had been observed.9b Subsequently, Spokoyny and co-workers reported the synthesis of an isomeric mixture of Pt complexes of [1,1′-bis(ortho-carborane)] with bipyridyl co-ligands;11c in one isomer the bis(carborane) was C,C′-bound and in the other it was C,B′-bound (subsequently he was able to prepare exclusively the C,C′-bound isomer by using a different synthetic strategy).11d Heating the mixture ‘under forcing conditions’ did not drive it to one isomer suggesting that the two isomers were formed via different pathways.
Thus, although it is potentially of great interest to be able to isomerise bis(carborane) from C,C′-bound to a metal centre to C,B′-bound under controlled conditions, no system has so far achieved this. We now describe the controlled isomerisation of a C,C′-bound bis(phosphine) ruthenium complex to its C,B′-bound isomer.
The room temperature reaction of [Ru(κ3-2,2′,3′-{1-(1′-closo-1′,2′-C2B10H10)-closo-1,2-C2B10H10})(p-cymene)] (I) with 5 equivalents of PMePh2 in THF produced a deep-red solution from which both red and yellow components were isolated by preparative thin-layer chromatography (TLC). Although the yellow product appeared stable to work-up, repeated chromatography of the red product (at room temperature) always afforded a small amount of the yellow species, implying that the red and yellow species were related as kinetically- and thermodynamically-stable isomers.
Repeating the reaction at 0 °C, eliminating the chromatographic work-up and crystallising the product at −20 °C allowed the red species (1) to be isolated in good yield (80%) in pure form.§ Mass spectrometry of 1 gave a molecular ion consistent with displacement of the p-cymene ligand of I by two PMePh2 ligands. Although the 11B{1H} NMR spectrum at −50 °C was largely uninformative the 1H spectrum revealed, in addition to multiplet resonances associated with the Ph groups, two doublets arising from the two PMePh2 units, implying the two phosphine ligands are inequivalent and confirmed by the presence of two mutual doublets, JPP = 28.3 Hz, in the 31P{1H} NMR spectrum. Importantly, the 1H{11B} NMR spectrum showed, in addition to resonances between 3 and −1 ppm associated with cage BHexo atoms, a doublet resonance at −3.27 ppm integrating for 1H and indicative of B–H⇀Ru (showing coupling to only one P atom). Notably absent from the 1H and 1H{11B} spectra of 1 was a resonance arising from cage CH.
Collectively these data suggest that in 1 the bis(ortho-carborane) unit is bound to the Ru atom in X2(C,C′)L mode, i.e. via both cage C atoms, unlike the situation in the previously isolated PPh3 and dppe analogues,9b and this was subsequently confirmed by crystallographic analysis (Fig. 1).¶
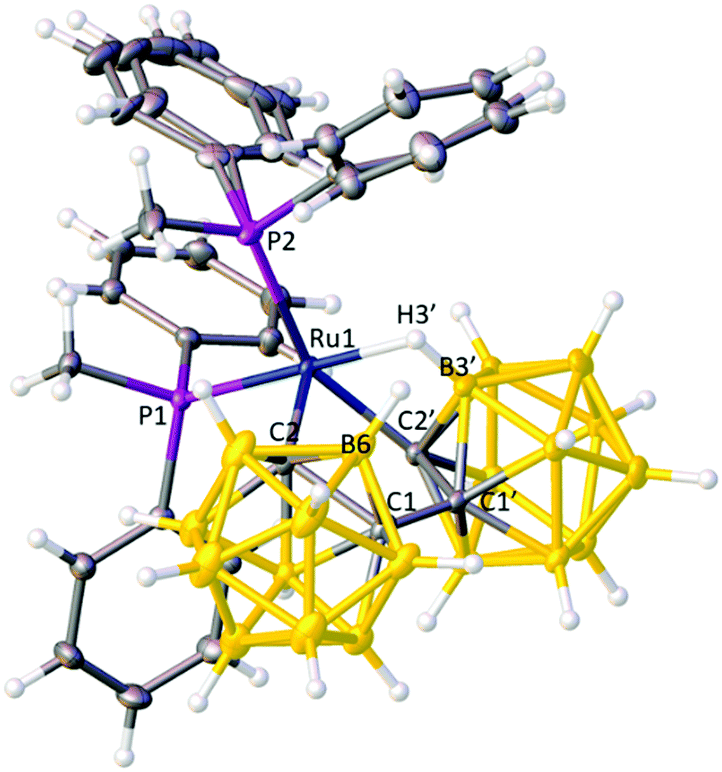 | ||
| Fig. 1 Structure of compound 1 (solvent omitted) with thermal ellipsoids drawn at the 50% probability level, except for H atoms. Both phenyl groups bound to P2 are partially disordered. Ru1–C2 2.0452(15), Ru1–C2′ 2.1472(14), Ru1–H3′ 1.872(17), Ru1–B3′ 2.3388(15), Ru1–P1 2.2769(3), Ru1–P2 2.3341(4), C1–C1′ 1.5113(19) Å. | ||
The bis(carborane) unit is indeed bonded to the metal atom via σ bonds from C2 and C2′ and a B3′–H3′⇀Ru B–agostic interaction; thus compound 1 is formulated as [Ru(κ3-2,2′,3′-{1-(1′-closo-1′,2′-C2B10H10)-closo-1,2-C2B10H10})(PMePh2)2]. The geometry at Ru is approximately square-pyramidal (C2 apex). The Ru–C2′ σ bond is particularly distorted, as evidenced by the angle Ru1–C2′–P ca. 134° cf. Ru1–C2–Q ca. 164° (P and Q are the centroids of the primed and unprimed icosahedra, respectively), presumably as a result of the need to accommodate C2′ and the B3′H3′ unit in two cis ligand positions. The Ru–C bond lengths are significantly different (shorter to C2), as are the Ru–P bond lengths (shorter to P1), in both cases reflecting the relative trans influences of the ligands (or vacant site) opposite.
Solutions of 1 slowly change from deep red to yellow in colour as the compound isomerises to a new species 2, a process easily followed by 31P{1H} NMR spectroscopy with two new higher-frequency doublets growing in at the expense of the original ones. At room temperature in CD2Cl2 the conversion is typically 15% after 6 h but is accelerated on heating (ca. 75% conversion after 2 h at 40 °C) and retarded on addition of excess phosphine (ca. 10% conversion after 24 h).
Compound 2 can be conveniently prepared in good yield (64%) by repeating the original synthesis at room temperature and then stirring for ca. 2 h at 40 °C followed by work-up involving column chromatography.|| Mass spectrometry is fully consistent with 2 being an isomer of compound 1. The 11B{1H} NMR spectrum of 2 is again relatively uninformative save that, as for 1, the chemical shifts imply a closo cage. The 1H NMR spectrum of 2 again reveals two sets of doublets for the methyl protons of the PMePh2 ligands and, additionally, an integral-1 resonance assigned to CcageH which unfortunately overlaps with the high-frequency component of one of the CH3 doublets (δ 1.85 ppm). However, further evidence for a cage {CH} unit derives from a resonance in the 13C NMR spectrum at δ 67.5 ppm assigned as CH by DEPT spectroscopy. The presence of a B–H⇀Ru interaction in 2 was established by the observation of an integral-1 doublet at −3.99 ppm in the 1H{11B} NMR spectrum.
Thus the NMR data imply an X2(C,B′)L bonding mode for the bis(ortho-carborane) unit in 2 as has previously been established for the PPh3 and dppe analogues,9b and this was subsequently confirmed by an X-ray diffraction study (Fig. 2).** Crystals of 1 and 2 (both studied as their 0.5CH2Cl2 solvates) are isomorphous and at a molecular level the two species differ only in the relative positions of a C and B atom in one cage. It was therefore imperative that cage vertices were correctly identified as either C or B in both crystallographic studies and this was established unambiguously by use of the VCD and BHD methods.15
 | ||
| Fig. 2 Structure of compound 2 (solvent omitted) with thermal ellipsoids drawn as in Fig. 1. The view is chosen to reflect similarity with the structure of compound 1. Ru1–C2 2.1375(13), Ru1–B3′ 2.0273(15), Ru1–H6 1.867(18), Ru1–B6 2.3265(14), Ru1–P3 2.2555(3), Ru1–P4 2.3153(3), C1–C1′ 1.5116(18) Å. | ||
The atomic numbering chosen for 2 reflects the likelihood that the B atom now σ-bonded to Ru (B3′) is the same atom which was involved in the B–agostic bond in 1, chemically-sensible in that the B3′–H3′ bond of 1 would be activated by such an interaction. In compound 2 the B–H⇀Ru interaction is now from the unprimed cage and involves B6H6 by conventional numbering. Thus, as compound 1 isomerises to compound 2 the essential changes may be summarised as; breaking of the B3′–H3′ bond and formation of a direct Ru–B3′ bond; breaking of the Ru–C2′ bond; formation of a new B6–H6⇀Ru bond; and net transfer of H from B3′ to C2′. These changes are summarised in Scheme 1, but at this stage we do not have any detailed mechanistic information. Compound 2 is therefore established as [Ru(κ3-2,3′,6-{1-(1′-closo-1′,2′-C2B10H10)-closo-1,2-C2B10H10})(PMePh2)2]. In compound 2 the two phosphine ligands again lie opposite the C–Ru σ bond and the B–agostic bond and have been labelled P3 and P4 to avoid any implied direct relationship to the phosphines in 1. Fully consistent with the structure of 1 there is considerable distortion of the Ru1–C2 bonding relative to the Ru1–B3′ bonding (Ru1–C2–R ca. 134° cf. Ru1–B3′–S ca. 158° where R and S are the centroids of the primed and unprimed icosahedra, respectively) and the relative lengths of the Ru–P bonds (shorter to P3) reflect the nature of the trans unit.
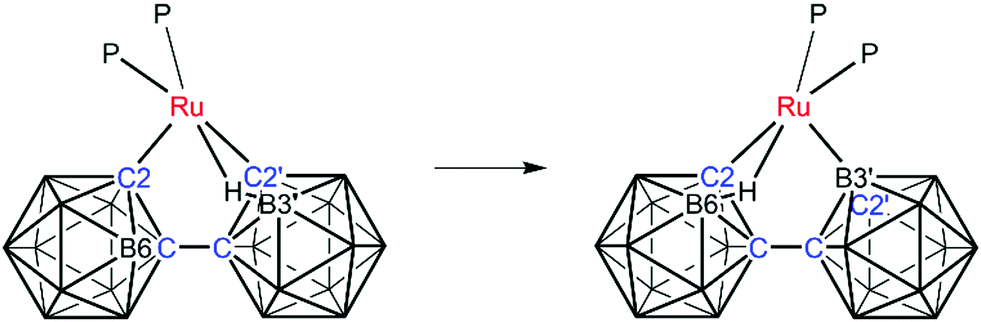 | ||
| Scheme 1 Suggested transformation of compound 1 (left) into compound 2 (right). | ||
In conclusion we have demonstrated the first controlled isomerisation of a bis(carborane) ligand from C,C′-bound to a metal centre to C,B′-bound, with no other change in the molecule. In the C,B′-bound isomer the metal centre will be relatively electron rich, and so this kind of isomerisation has the potential to allow the properties of the molecule, including catalytic properties, to be tuned in a controlled way.
We thank the Engineering & Physical Sciences Research Council and the CRITICAT Centre for Doctoral Training for a PhD studentship awarded to R. J. J. (grant no. EP/L016419/1). We also thank Prof. S. A. Macgregor for many helpful discussions.
Conflicts of interest
There are no conflicts to declare.Notes and references
- R. N. Grimes, Carboranes, Elsevier, Amsterdam, The Netherlands, 3rd edn, 2016 Search PubMed.
- M. F. Hawthorne, D. C. Young, T. D. Andrews, D. V. Howe, R. L. Pilling, A. D. Pitts, M. Reintjes, L. F. Warren Jnr and P. A. Wegner, J. Am. Chem. Soc., 1968, 90, 879–896 CrossRef CAS.
- D. M. P. Mingos, M. I. Forsyth and A. J. Welch, J. Chem. Soc., Dalton Trans., 1978, 1363–1374 RSC.
- G. G. Hlatky, H. W. Turner and R. R. Eckman, J. Am. Chem. Soc., 1989, 111, 2728–2729 CrossRef CAS.
- S. A. Brew, J. C. Jeffery, M. D. Mortimer and F. G. A. Stone, J. Chem. Soc., Dalton Trans., 1993, 1365–1374 Search PubMed.
- C. B. Knobler, T. B. Marder, E. A. Mizusawa, R. G. Teller, J. A. Long, P. E. Behnken and M. F. Hawthorne, J. Am. Chem. Soc., 1984, 106, 2990–3004 CrossRef CAS.
- N. Bresciani, M. Calligaris, P. Delise, G. Nardin and L. Randaccio, J. Am. Chem. Soc., 1974, 96, 5642–5643 CrossRef CAS.
- I. A. Lobanova, V. I. Bregadze, S. V. Timofeev, P. V. Petrovskii, Z. A. Starikova and F. M. Dolgushin, J. Organomet. Chem., 2000, 597, 48–53 CrossRef CAS.
- (a) R. A. Love and R. Bau, J. Am. Chem. Soc., 1972, 94, 8274–8276 CrossRef CAS; (b) L. E. Riley, A. P. Y. Chan, J. Taylor, W. Y. Man, D. Ellis, G. M. Rosair, A. J. Welch and I. B. Sivaev, Dalton Trans., 2016, 45, 1127–1137 RSC.
- (a) Z. Zheng, M. Diaz, C. B. Knobler and M. F. Hawthorne, J. Am. Chem. Soc., 1995, 117, 12338–12339 CrossRef CAS; (b) F. Teixidor, R. Núñez, C. Viñas, R. Sillanpää and R. Kivekäs, Angew. Chem., Int. Ed., 2000, 39, 4290–4292 CrossRef CAS; (c) F. Teixidor, G. Barberà, A. Vaca, R. Kivekäs, R. Sillanpää, J. Oliva and C. Viñas, J. Am. Chem. Soc., 2005, 127, 10158–10159 CrossRef CAS.
- (a) A. M. Spokoyny, C. W. Machan, D. J. Clingerman, M. S. Rosen, M. J. Wiester, R. D. Kennedy, C. L. Stern, A. A. Sarjeant and C. A. Mirkin, Nat. Chem., 2011, 3, 590–596 CrossRef CAS PubMed; (b) A. M. Spokoyny, C. D. Lewis, G. Teverovskiy and S. L. Buchwald, Organometallics, 2012, 31, 8478–8481 CrossRef CAS; (c) K. O. Kirlikovali, J. C. Axtell, A. Gonzalez, A. C. Phung, S. I. Khan and A. M. Spokoyny, Chem. Sci., 2016, 7, 5132–5138 RSC; (d) K. O. Kirlikovali, J. C. Axtell, K. Anderson, P. I. Djurovich, A. L. Rheingold and A. M. Spokony, Organometallics, 2018, 37, 3122–3131 CrossRef CAS.
- Recent publications: (a) I. B. Sivaev and V. I. Bregadze, Coord. Chem. Rev., 2019, 392, 146–176 CrossRef CAS and references therein ; (b) S. Yruegas, J. C. Axtell, K. O. Kirlikovali, A. M. Spokoyny and C. D. Martin, Chem. Commun., 2019, 55, 2892–2895 RSC; (c) J. Wu, K. Cao, C.-Y. Zhang, T.-T. Xu, L.-F. Ding, B. Li and J. Yang, Org. Lett., 2019, 21, 5986–5989 CrossRef CAS; (d) R. J. Jeans, A. P. Y. Chan, L. E. Riley, J. Taylor, G. M. Rosair, A. J. Welch and I. B. Sivaev, Inorg. Chem., 2019, 58, 11751–11761 CrossRef CAS; (e) A. Benton, D. J. Durand, Z. Copeland, J. D. Watson, N. Fey, S. M. Mansell, G. M. Rosair and A. J. Welch, Inorg. Chem., 2019, 58, 14818–14829 CrossRef CAS; (f) I. Chambrier, D. L. Hughes, R. J. Jeans, A. J. Welch, P. H. M. Budzelaar and M. Bochmann, Chem. – Eur. J., 2020, 26, 939–947 CrossRef CAS PubMed; (g) A. P. Y. Chan, J. A. Parkinson, G. M. Rosair and A. J. Welch, Inorg. Chem., 2020, 59, 2011–2023 CrossRef CAS; (h) A. P. Y. Chan, G. M. Rosair and A. J. Welch, Molecules, 2020, 25, 519 CrossRef CAS; (i) A. J. Welch, Structure and Bonding, Springer, Berlin, Heidelberg, 2021 DOI:10.1007/430_2020_80.
- (a) M. F. Hawthorne and D. A. Owen, J. Am. Chem. Soc., 1971, 93, 873–880 CrossRef CAS; (b) D. E. Harwell, J. McMillan, C. B. Knobler and M. F. Hawthorne, Inorg. Chem., 1997, 36, 5951–5955 CrossRef CAS.
- (a) M. J. Martin, W. Y. Man, G. M. Rosair and A. J. Welch, J. Organomet. Chem., 2015, 798, 36–40 CrossRef CAS; (b) Z.-J. Yao, Y.-Y. Zhang and G.-X. Jin, J. Organomet. Chem., 2015, 798, 274–277 CrossRef CAS.
- (a) A. McAnaw, G. Scott, L. Elrick, G. M. Rosair and A. J. Welch, Dalton Trans., 2013, 42, 645–664 RSC; (b) A. McAnaw, M. E. Lopez, D. Ellis, G. M. Rosair and A. J. Welch, Dalton Trans., 2014, 43, 5095–5105 RSC; (c) A. J. Welch, Crystals, 2017, 7, 234 CrossRef.
Footnotes |
| † Dedicated with very best wishes to Professors Francesc Teixidor and Clara Viñas, colleagues and good friends for more than 30 years, on the occasion of their 70th birthdays. |
| ‡ Electronic supplementary information (ESI) available. CCDC 2117898 and 2117899. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1cc06119d |
| § NMR data for 1 (CD2Cl2, −50 °C): 11B{1H}; δ 14 to −22 overlapping resonances with prominent maxima at −3.7, −7.9 (assume 20B). 1H; δ 7.51–7.45 [m, 2H, PCH3(C6H5)2], 7.44–7.38 [m, 1H, PCH3(C6H5)2], 7.36–7.23 [m, 7H, PCH3(C6H5)2], 7.21–7.09 [m, 6H, PCH3(C6H5)2], 7.06–6.99 [m, 2H, PCH3(C6H5)2], 6.96–6.89 [m, 2H, PCH3(C6H5)2], 2.35, [d, JHP = 7.9 Hz, 3H, PCH3(C6H5)2], 2.16 [d, JHP = 10.0 Hz, 3H, PCH3(C6H5)2]. 1H{11B} as for 1H plus δ 2.67 to 2.08 considerable overlap of BH and CH3 resonances (total integral 9BH + 6CH) with prominent BH maxima at 2.61, 2,46 and 2.24, 1.89 (4H), 1.75 to 1.47 overlapping resonances with prominent maxima at 1.70, 1.67, 1.60 and 1.55 (total integral 5H, BH), 0.95 (1H, BH), −3.27 (d, JHP = 31.0 Hz, 1H, BHRu). 31P{1H}; δ 34.3 (d, JPP = 28.3 Hz, 1P), 22.6 (d, JPP = 28.3 Hz, 1P). EIMS: envelope centred on m/z 786.3 (M+). |
| ¶ Crystal data for 1·0.5CH2Cl2: C30.5H47B20ClP2Ru, M = 828.34, monoclinic, C2/c, a = 38.4205(9), b = 10.5927(2), c = 21.1877(5) Å, β = 111.3870(10)°, V = 8029.1(3) Å3, F(000) = 3368.0 e, Dcalc = 1.371 Mg m−3, Cu-Kα X-radiation, λ = 1.54178 Å, μ = 4.693 mm−1, of 54551 data measured to θmax = 74.78° on a Bruker D8 Venture diffractometer, 8168 were unique and were used to solve and refine the structure to R = 0.0213, wR2 = 0.0529, CCDC 2117898‡. |
| || NMR data for 2 (CD2Cl2, room temperature): 11B{1H}; δ 0.2 (2B), −1.9 to −15.9 (overlapping resonances with maxima at −4.6, −8.7, and −14.6, total integral 17B), −17.0 (1B). 1H; δ 7.60–7.55 [m, 2H, PCH3(C6H5)2], 7.43–7.20 [m, 18H, PCH3(C6H5)2], 2.34 [d, JPH = 7.9 Hz, 3H, PCH3(C6H5)2], 1.85 (s, 1H, CcageH) overlapping with 1.84 [d, JPH = 9.5 Hz, 3H, PCH3(C6H5)2]. 1H{11B}; as for 1H plus δ 2.53 (2H, BH), 2.50 (1H, BH), 2.38 (1H, BH), 2.26 (3H, BH), 2.19 (1H, BH), 2.17 (2H, BH), 2.10 (2H, BH), 2.00 (1H, BH), 1.96 (1H, BH), 1.76 (1H, BH), 1.58 (3H), BH), −3.99 (d, JHP = 27.6 Hz, 1H, BHRu). 13C; δ 132.4 (d, JCP = 11.3 Hz, Carom.H), 132.1 (d, JCP = 10.5 Hz, Carom.H), 131.8 (d, JCP = 11.2 Hz, Carom.H), 131.5 (d, JCP = 9.7 Hz, Carom.H), 130.8 (d, JCP = 2.2 Hz, Carom.H), 130.4 (d, JCP = 2.0 Hz, Carom.H), 130.3 (d, JCP = 2.5 Hz, Carom.H), 130.2 (d, JCP = 1.9 Hz, Carom.H), 129.3 (d, JCP = 9.7 Hz, Carom.H), 129.1 (d, JCP = 9.3 Hz, Carom.H), 128.9 (d, JCP = 9.3 Hz, Carom.H), 128.7 (d, JCP = 9.6 Hz, Carom.H), 91.8 (C), 77.8 (C), 67.5 (CcageH), 18.8 (d, JCP = 28.2 Hz, CH3), 14.2 (d, JCP = 34.6 Hz, CH3). 31P{1H}; δ 41.5 (d, JPP = 28.0 Hz, 1P), 28.0 (d, JPP = 28.0 Hz, 1P). EIMS: envelope centred on m/z 786.3 (M+), 586.2 (M+ − PMePh2). |
| ** Crystal data for 2·0.5CH2Cl2: C30.5H47B20ClP2Ru, M = 828.34, monoclinic, C2/c, a = 38.0931(9), b = 10.6717(3), c = 21.3413(5) Å, β = 111.8580(10)°, V = 8051.9(4) Å3, F(000) = 3368.0 e, Dcalc = 1.367 Mg m−3, Cu-Kα X-radiation, λ = 1.54178 Å, μ = 4.680 mm−1, 64154 data to θmax = 74.73°, 8221 unique, R = 0.0208, wR2 = 0.0512, CCDC 2117899‡. |
| This journal is © The Royal Society of Chemistry 2022 |