
ROS activated prodrug for ALDH overexpressed cancer stem cells†
Miae
Won‡
 ,
Ji Hyeon
Kim‡
,
Myung Sun
Ji‡
and
Jong Seung
Kim
*
,
Ji Hyeon
Kim‡
,
Myung Sun
Ji‡
and
Jong Seung
Kim
*
Department of Chemistry, Korea University, Seoul 02841, Korea. E-mail: jongskim@korea.ac.kr
First published on 7th December 2021
Abstract
Aldehyde dehydrogenase (ALDH), a cancer stem cell biomarker, is related to drug resistance. Co-treatment of anti-cancer drug (CPT) and ALDH inhibitor (DEAB) can overcome the drug resistance of cancer stem cells (CSCs) and finally cure cancers without relapse. We herein introduce a prodrug (DE-CPT) – consisting of 1,3-oxathiolane as an ROS responsive scaffold, and an aldehyde protecting group of DEAB – to deliver the CPT and DEAB upon reaction with ROS. From tests of the sphere-forming ability and CSC marker subpopulation, we found that DE-CPT efficiently decreases the CSCs population and kills the cancer cells.
Although nanoparticles and small molecules for cancer treatment have been actively developed, cancer recurrence and metastasis are still issues.1,2 Since the first identification of cancer stem cells (CSCs) in the 1990s, attention toward CSCs in the cancer research field has been heightened because cancer can be caused by even a small population of CSCs.3,4 The CSCs have stem cell characteristics such as self-renewal and differentiation capacity.5,6 Further, due to the multidrug resistance property of CSCs, they can survive even after anti-cancer agent treatment, causing cancer recurrence and metastasis. Eradicating CSCs therefore is one of the most challenging strategies in cancer therapy because the CSCs have a crucial role in tumour formation, metastasis and recurrence.7
Aldehyde dehydrogenase (ALDH) is an enzyme that converts a toxic aldehyde to a carboxylic acid and its overexpression is considered a representative CSC biomarker.8–10 Recent studies have demonstrated that high ALDH activity is associated with resistance of CSCs to chemotherapeutic drugs, and thus ALDH inhibitors are recommended to be co-treated with anticancer drugs.11–13 However, treatment of anti-cancer agents with ALDH inhibitor can be rather risky because even normal stem cells share high ALDH activity with CSCs, so normal stem cells can be damaged as well.14 Therefore, a precisely designed prodrug system for the use of ALDH inhibitor that can be selectively activated in the tumour tissue and not normal cells, is required to circumvent side effects.
Representative factors of the tumour microenvironment (TME) are hypoxic conditions, increased reactive oxygen species (ROS) levels, and low pH. Using these features, drug delivery systems (DDS) able to activate in specific tumour environments have been well developed.15,16 Along with the development of diverse ROS-responsive linkers,17,18 ROS such as hydrogen peroxide (H2O2) have emerged as one of the important triggering tools conferring cancer therapeutic selectivity.19–21
Given the above considerations, we have developed a novel ROS activatable prodrug consisting of ALDH inhibitor and anti-cancer drug, able to eradicate cancer cells and CSCs simultaneously. The selected ALDH inhibitor in the current study is 4-diethylaminobenzaldehyde (DEAB), commonly used in ALDH related assays such as the Aldefluor assay which is a well-established tool to measure the ALDH activity in CSCs.22,23 However, the highly reactive aldehyde functional group in DEAB was found to be unstable in physiological systems.24,25 For this stability issue, the aldehyde functional group of DEAB can be covered by a unique ROS trigger, 1,3-oxathiolane, which is a well-known aldehyde protecting group and can be deprotected by hypochlorous acid (HOCl).26,27 Moreover, it was also reported that 1,3-oxathiolane can release the aldehyde in H2O2 conditions.28,29 In regard to selectively targeting CSCs over normal cells with synergistic chemotherapy, we have designed an ALDH targeting prodrug DE-CPT, having H2O2 responsive 1,3-oxathiolane moiety. Overall, the DE-CPT can be activated by ROS to simultaneously release ALDH inhibitor (DEAB) and anticancer agent (CPT) as demonstrated in Fig. 1.
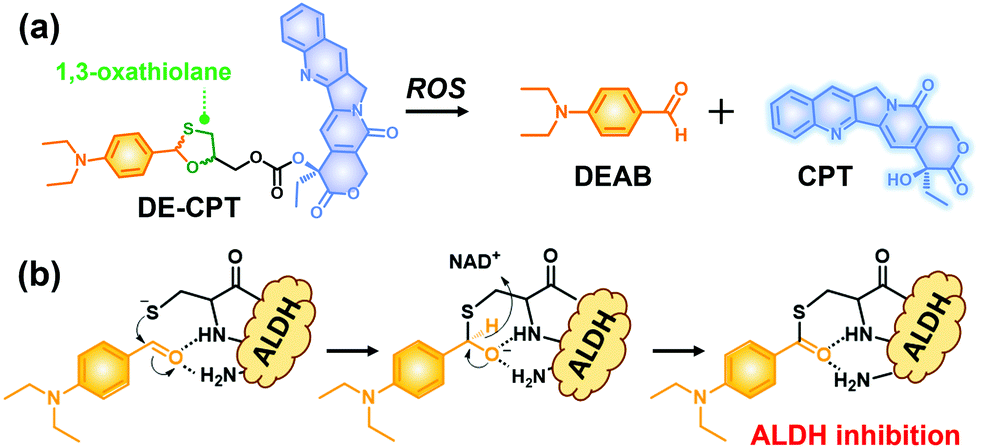 | ||
| Fig. 1 (a) Chemical structure of DE-CPT and ROS responsive reaction to release free DEAB and CPT. (b) ALDH inhibition mechanism of released DEAB. | ||
DE-CPT was successfully prepared as shown in Scheme S1 (ESI†), and its full characterization data (1H and 13C NMR spectroscopy and ESI-MS) are given in the ESI† (Fig. S1–S31). Briefly, reaction of α-thioglycerol with DEAB to protect aldehyde gave 1,3-oxathiolane. Next, the free hydroxyl functional group was reacted with p-nitrophenyl chloroformate for further modification. Finally, intermediate 2 was reacted with CPT to provide DE-CPT.
The proposed activation mechanism of DE-CPT is shown in Fig. 2a. First of all, sulfur in 1,3-oxathiolane of DE-CPT is oxidized by ROS followed by the release of deprotected DEAB. In the intermediate sulfonic acid molecule, the remaining hydroxy group attacks the adjacent carbonate functional group followed by cyclization reaction to release the CPT drug. The release of DEAB is monitored by time-dependent absorbance spectrum, spotting the increment of absorbance at 360 nm (Fig. 2b and Fig. S11a, ESI†).23 The release of free CPT was also monitored by fluorescence enhancement at 435 nm (Fig. 2c and d). Based on the ROS activation mechanism, we noticed that DEAB and CPT were released from the DE-CPT under H2O2 conditions, whereas no change of spectra was observed in the absence of H2O2. We could validate the reaction completion from the full release of DEAB and CPT by HPLC assay (Fig. 2e and Fig. S11b, ESI†). The DE-CPT peak disappears within 2 h, while the DEAB and the intermediate peaks emerge with a small amount of CPT indicates that the DEAB release completes before CPT release. We also noticed that the intermediate peak disappears within 10 h. So, it could therefore be assumed that the intermediate is a sulfonic acid molecule containing CPT. We further verified the DE-CPT reactivity under mildly acidic conditions (pH 6.5). Under these conditions, although the reactivity was slightly decreased, both DEAB and CPT can be released (Fig. S12 and S13, ESI†). Based on the reactivity toward H2O2, selectivity studies of DE-CPT were performed to determine the interference by other analytes such as amino acid and metal ions. As shown in Fig. S14 (ESI†), the H2O2 could exclusively change absorbance and fluorescence. Although small increments of fluorescence were observed in the case of highly nucleophilic GSH and cysteine, their change in fluorescence was negligible compared to the use of H2O2. Next, we examined the reactivity of DE-CPT toward HOCl. Compared to H2O2, a relatively small quantity of HOCl could release considerable amounts of both DEAB and camptothecin (Fig. S15, ESI†). This result indicates that DE-CPT also can react under HOCl, another important ROS in biological systems.
 | ||
Fig. 2 (a) Mode of action of DE-CPT under abundant ROS in cancer cells. (b) Absorbance spectra of DE-CPT (10 μM) treated with 100 eq. of H2O2. (c) Fluorescence spectra of DE-CPT (10 μM) treated with 100 eq. of H2O2. The solution was incubated for up to 3 h at 37 °C in ACN/PBS (pH 7.4) buffer (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3) solution. (d) The fluorescence changes of DE-CPT at 435 nm determined from data in (c) and Fig. S11(b), ESI.† (e) HPLC analysis (absorbance at 254) DE-CPT incubated with 100 eq. H2O2 at 37 °C for 10 h. Also shown are the chromatograms of DEAB and CPT. :3) solution. (d) The fluorescence changes of DE-CPT at 435 nm determined from data in (c) and Fig. S11(b), ESI.† (e) HPLC analysis (absorbance at 254) DE-CPT incubated with 100 eq. H2O2 at 37 °C for 10 h. Also shown are the chromatograms of DEAB and CPT. | ||
To offer further supportive evidence that DE-CPT can be activated through the thiol oxidation, ESI-MS spectra were taken after 6 h reaction of DE-CPT with H2O2 (Fig. S16, ESI†). [M + H]+ peak of released DEAB (m/z: 178.20) was detected in positive scan mode. In negative scan mode, [M − H]− peak of the released camptothecin (CPT) (m/z: 346.95) was seen, corresponding to the results of absorption and emission spectra we have observed. Essentially, the two desired peaks, [M − H]− peak of the intermediate (m/z: 582.95) and the cyclization reaction by-product (m/z: 180.90), were also identified (Fig. S16, ESI†). These results coincide well with the proposed mechanism shown in Fig. 2a, where DE-CPT is activated through the thiol oxidation.
Encouraged by the fact that our rationally designed DE-CPT can release DEAB and CPT in H2O2 solution, the cell-based in vitro experiments were next performed. Cell viability of DE-CPT was firstly conducted to evaluate if this prodrug system efficiently works in cancer cells. We observed promising results of cancer cell selectivity of DE-CPT when we compare the cell viability of DE-CPT with that of CPT. It is noteworthy that DE-CPT causes minor damage to normal cells (MCF10A) compared to CPT at the same concentration (Fig. 3a). On the contrary, DE-CPT was found to be more toxic to breast cancer cells (BT474 and MDA-MB-231) than CPT treated cells at the same concentration (Fig. 3b and Fig. S17, ESI†, respectively).
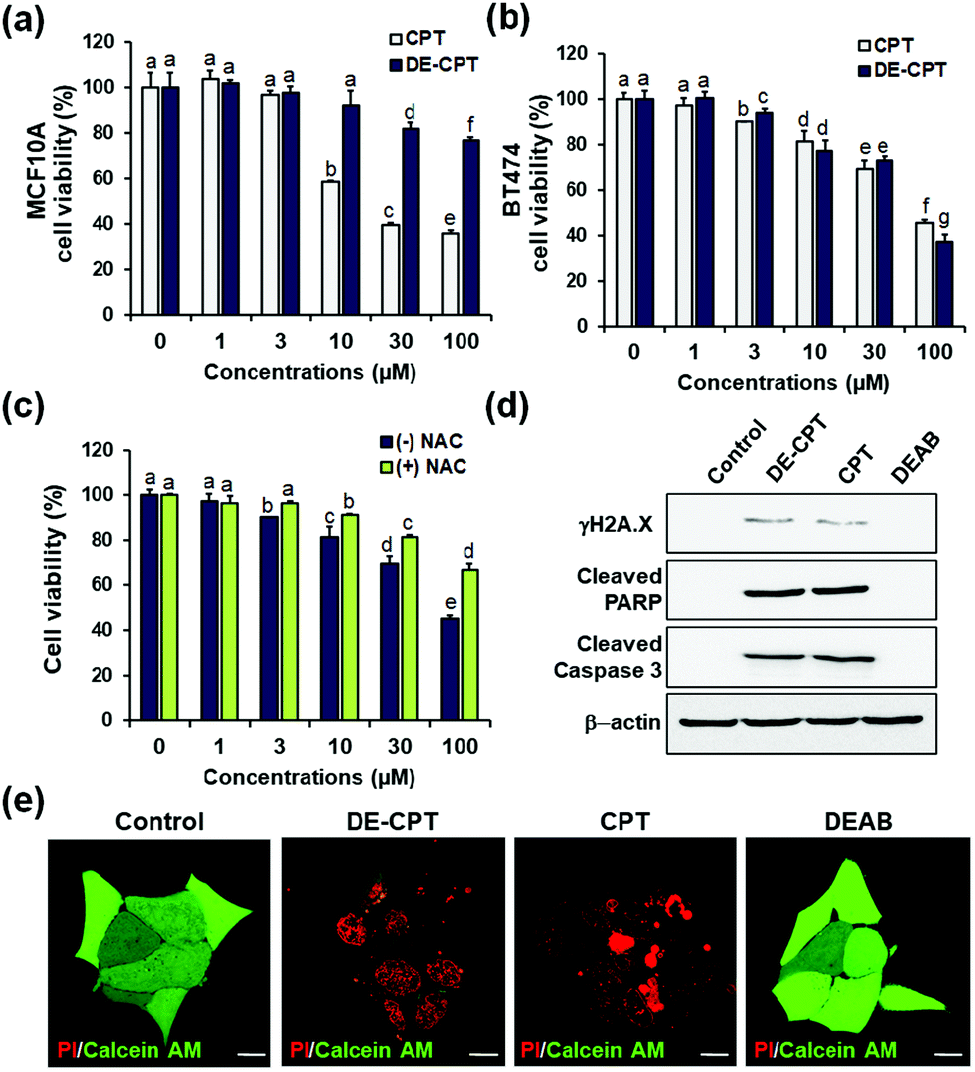 | ||
| Fig. 3 (a and b) Cell viability of DE-CPT in MCF10A and BT474 cells. (c) Cell viability of DE-CPT in the absence or presence of a ROS quencher (NAC) in BT-474 cells. Data are represented as mean ± SEM (n = 3). Statistical significance was determined by a two-way ANOVA test with a post-hoc Bonferroni test. The different letters (e.g., a–g) signify datasets that are statistically distinct (p < 0.05). (d) Cell death signaling process of DE-CPT in BT474 cells. (e) Fluorescence images of PI/calcein AM stained BT474 cells treated with control (1% DMSO), DE-CPT, CPT and DEAB for 48 h. Scale bar = 50 μm. | ||
To verify that DE-CPT activation is caused by endogenous ROS in cancer cells, cells were pre-treated with ROS scavenger N-acetylcysteine (NAC) (Fig. 3c). NAC-treated cells show significant attenuation in cell cytotoxicity compared to NAC-untreated cells, demonstrating that DE-CPT activation is due to a high level of ROS in cancer cells. At the same time, DEAB is not toxic even at a high concentration because it is an inhibitor, not a chemotherapeutic drug (Fig. S18, ESI†). These results collectively suggest that ROS in cancer cells activate the DE-CPT which releases the DEAB to inhibit the ALDH, thereby making the cancer cells sensitive to CPT. To explore apoptotic cell death signalling, we next performed western blotting analysis for γH2A.X, cleaved poly ADP-ribose polymerase (PARP), and cleaved caspase3 protein expression. We found that the apoptosis-inducing ability of DE-CPT is similar to that of CPT from the results of high levels of cleaved PARP and phosphorylated H2A.X (Fig. 3d and Fig. S19, ESI†). These are marker proteins for the DNA damage repair of cancer cells and CSCs. Caspase-dependent downstream cascade (cleaved caspase-3) accumulation indicated apoptotic cell death signalling as well.30,31 To further verify the cellular damage, BT474 cells were co-stained with PI and calcein-AM after incubation with 30 μM DE-CPT for 48 h. As shown in fluorescence imaging using confocal laser scanning microscopy (CLSM), in Fig. 3e, the solid red region indicates dead cells in DE-CPT and CPT with weak calcein-AM signals in the live cells.
Given that DE-CPT is a prodrug of ALDH inhibitor-conjugated anti-cancer drug, we focused on the therapeutic effects reducing the cancer stem cell characteristics. CSCs are considered to cause tumorigenesis to form three-dimensional spheroids.32 Sphere-forming efficiency (SFE) is often evaluated to determine the removal degree of CSCs. BT474 cells were seeded in ultra-low attached plates using the CSC medium, and treated with DE-CPT, CPT, DEAB or control (1% DMSO) on the second day. After 5 days, the sphere morphologies and the number of retained spheres having a size over 150 μm were counted. Compared to the CPT treated group, DE-CPT (30 μM) treated cells showed decreased spheroid-forming ability with almost complete damage (>95%) as shown in Fig. 4a and b. These results provide robust evidence for the suppression of the CSC population. Next, ALDH positive fraction in BT474 adhesive cells and spheroid cells was quantified by flow cytometry. Compared to the control (1% DMSO) and CPT treated groups, the ALDH positive subpopulation was dramatically decreased by DE-CPT (Fig. 4c). This coincides with a significant reduction in two typical breast CSC biomarkers, i.e., ALDH1 and CD44, indicative of CSC enrichment (p < 0.05, Fig. 4d and Fig. S20 and S21, ESI†).33 We therefore conclude that the results account for the inhibition of ALDH by released DEAB to reduce the stem cell-expressing CSC population, which can evidently explain why the CSC-killing profile of DE-CPT is superior to that of CPT itself.
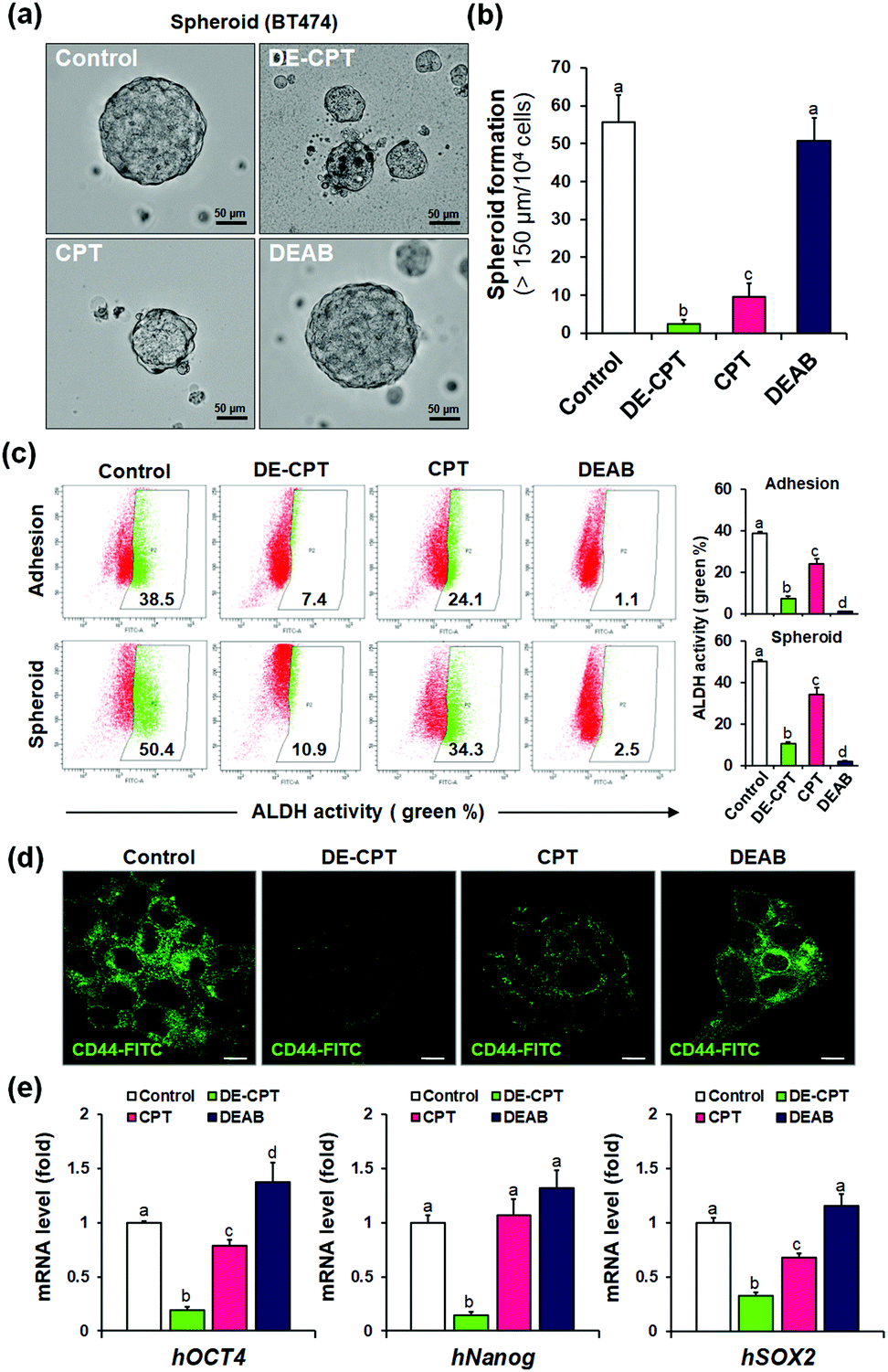 | ||
| Fig. 4 (a) Spheroid ability by DE-CPT. Scale bar = 50 μm. (b) Spheroid formation of DE-CPT. (c) ALDH1 activity in adhesion or spheroid of BT474 cells by flow cytometry. (d) Fluorescence images of CD44 (stained with FITC anti-CD 44) of DE-CPT. Scale bar = 50 μm. (e) mRNA levels of CSC markers of human OCT4, Nanog, Sox2 in BT474 tumour spheroids treated with 30 μM DE-CPT. Data are represented as mean ± SEM (n = 3 in D). Statistical significance was determined by a one-way ANOVA test with a post-hoc Bonferroni test. Different letters (e.g., a–d) signify datasets that are statistically distinct (p < 0.05). | ||
In parallel, to explore the suppression of CSC ability by activated DE-CPT, stem-cell marker gene of mRNA expressions were investigated. Human Oct4, Nanog, and Sox2 are the representative CSC expressing genes and are responsible for the drug resistance, proliferation, and metastasis of CSCs.34,35 A remarkable decrease of three stem cell-related gene markers was confirmed in DE-CPT treated spheroids in comparison with other treated groups. Taken together, although the CPT was as toxic to adhesive cells as DE-CPT (Fig. 3b), it has little effect on the reduction of the CSCs fraction. On the other hand, DE-CPT showed a significant impact in reducing the CSC population over CPT, a commercialized chemotherapeutic agent. These findings thereby further confirmed the success of our strategy and indicate that DE-CPT elicits anti-CSC efficacy via the induction of apoptosis, caspase activation and DNA damage by ROS activation in CSCs.
In summary, we have developed ALDH targeting prodrug DE-CPT, bearing H2O2 responsive 1,3-oxathiolane moiety, which can be activated by ROS to simultaneously release both the ALDH inhibitor and CPT. The ALDH targeting DE-CPT showed a clear advantage in selectivity toward cancer cells over normal cells. The synergistic effect by concomitant drug release of ALDH inhibitor (DEAB) and anti-cancer drug (CPT) induced an optimized cancer cell apoptosis. Moreover, we found that the DE-CPT can remarkably shrink the CSC fraction by decreasing the ALDH positive fraction, CSC marker genes, and representative surface marker CD44, whereas that in CPT-treated cells is insignificant. Therefore, we expect that the ALDH targeting prodrug DE-CPT that we have developed, provides a promising strategy for therapeutic approaches towards both cancer cell and cancer stem cell treatment.
This work was supported by the National Research Foundation of Korea (NRF) funded by the Ministry of Science and ICT (CRI project no. 2018R1A3B1052702 and no. 2019M3E5D1A01068998, J. S. K.) and the global PhD fellowship (GPF) program (no. 2019H1A2A1074096, J. H. K.), funded by the Ministry of Education. This research was supported by the Korea University Graduate School Junior Fellow Research Grant (J. H. K).
Conflicts of interest
There are no conflicts to declare.Notes and references
- S. D. Steichen, M. Caldorera-Moore and N. A. Peppas, Eur. J. Pharm. Sci., 2013, 48, 416–427 CrossRef CAS PubMed.
- J. Rautio, N. A. Meanwell, L. Di and M. J. Hageman, Nat. Rev. Drug Discovery, 2018, 17, 559–587 CrossRef CAS.
- T. Lapidot, C. Sirard, J. Vormoor, B. Murdoch, T. Hoang, J. Caceres-Cortes, M. Minden, B. Paterson, M. A. Caligiuri and J. E. Dick, Nature, 1994, 367, 645–648 CrossRef CAS.
- S. Bomken, K. Fiser, O. Heidenreich and J. Vormoor, Br. J. Cancer, 2010, 103, 439–445 CrossRef CAS PubMed.
- H. Clevers, Nat. Med., 2011, 17, 313–319 CrossRef CAS.
- E. Batlle and H. Clevers, Nat. Med., 2017, 23, 1124–1134 CrossRef CAS.
- A. Desai, Y. Yan and S. L. Gerson, Stem Cells Transl. Med., 2019, 8, 75–81 CrossRef PubMed.
- P. Marcato, C. A. Dean, C. A. Giacomantonio and P. W. Lee, Cell Cycle, 2011, 10, 1378–1384 CrossRef CAS.
- V. Muralikrishnan, T. D. Hurley and K. P. Nephew, Cancers, 2020, 12, 961 CrossRef CAS.
- X. Xu, S. Chai, P. Wang, C. Zhang, Y. Yang, Y. Yang and K. Wang, Cancer Lett., 2015, 369, 50–57 CrossRef CAS PubMed.
- T. Tanei, K. Morimoto, K. Shimadzu, S. J. Kim, Y. Tanji, T. Taguchi, Y. Tamaki and S. Noguchi, Clin. Cancer Res., 2009, 15, 4234–4241 CrossRef CAS PubMed.
- A. K. Croker and A. L. Allan, Breast Cancer Res. Treat., 2012, 133, 75–87 CrossRef CAS PubMed.
- R. Januchowski, K. Wojtowicz and M. Zabel, Biomed. Pharmacother., 2013, 67, 669–680 CrossRef CAS.
- C. Ginestier, M. H. Hur, E. Charafe-Jauffret, F. Monville, J. Dutcher, M. Brown, J. Jacquemier, P. Viens, C. G. Kleer, S. Liu, A. Schott, D. Hayes, D. Birnbaum, M. S. Wicha and G. Dontu, Cell Stem Cell, 2007, 1, 555–567 CrossRef PubMed.
- B. Chen, W. Dai, B. He, H. Zhang, X. Wang, Y. Wang and Q. Zhang, Theranostics, 2017, 7, 538–558 CrossRef CAS.
- A. Sharma, J. F. Arambula, S. Koo, R. Kumar, H. Singh, J. L. Sessler and J. S. Kim, Chem. Soc. Rev., 2019, 48, 771–813 RSC.
- J. P. Cadahía, V. Previtali, N. S. Troelsen and M. H. Clausen, Med. Chem. Commun., 2019, 10, 1531–1549 RSC.
- P. T. Wong and S. K. Choi, Chem. Rev., 2015, 115, 3388–3432 CrossRef CAS.
- H. Hagen, P. Marzenell, E. Jentzsch, F. Wenz, M. R. Veldwijk and A. Mokhir, J. Med. Chem., 2012, 55, 924–934 CrossRef CAS.
- G. Saravanakumar, J. Kim and W. J. Kim, Adv. Sci., 2017, 4, 1600124 CrossRef.
- F. Weinberg, N. Ramnath and D. Nagrath, Cancers, 2019, 11, 1191 CrossRef CAS.
- V. Koppaka, D. C. Thompson, Y. Chen, M. Ellermann, K. C. Nicolaou, R. O. Juvonen, D. Petersen, R. A. Deitrich, T. D. Hurley and V. Vasiliou, Pharmacol. Rev., 2012, 64, 520–539 CrossRef CAS PubMed.
- C. A. Morgan, B. Parajuli, C. D. Buchman, K. Dria and T. D. Hurley, Chem. – Biol. Interact., 2015, 234, 18–28 CrossRef CAS PubMed.
- S. J. Dylla, L. Beviglia, I. K. Park, C. Chartier, J. Raval, L. Ngan, K. Pickell, J. Aguilar, S. Lazetic, S. Smith-Berdan, M. F. Clarke, T. Hoey, J. Lewicki and A. L. Gurney, PLoS One, 2008, 3, e2428 CrossRef PubMed.
- M. I. Mahmoud, J. J. Potter, O. M. Colvin, J. Hilton and E. Mezey, Alcohol.: Clin. Exp. Res., 1993, 17, 1223–1227 CrossRef CAS.
- J. Han, M. Won, J. H. Kim, E. Jung, K. Min, P. Jangili and J. S. Kim, Chem. Soc. Rev., 2020, 49, 7856–7878 RSC.
- L. Yuan, L. Wang, B. K. Agrawalla, S. J. Park, H. Zhu, B. Sivaraman, J. Peng, Q. H. Xu and Y. T. Chang, J. Am. Chem. Soc., 2015, 137, 5930–5938 CrossRef CAS PubMed.
- J. R. Cashman, J. Proudfoot, Y. K. Ho, M. S. Chin and L. D. Olsen, J. Am. Chem. Soc., 1989, 111, 4844–4852 CrossRef CAS.
- S. P. Chavan, S. W. Dantale, K. Pasupathy, R. B. Tejwani, S. K. Kamat and T. Ravindranathan, Green Chem., 2002, 4, 337–338 RSC.
- S. Elmore, Toxicol. Pathol., 2007, 35, 495–516 CrossRef CAS.
- D. R. McIlwain, T. Berger and T. W. Mak, Cold Spring Harbor Perspect. Biol., 2013, 5, a008656 Search PubMed.
- H. F. Bahmad, K. Cheaito, R. M. Chalhoub, O. Hadadeh, A. Monzer, F. Ballout, A. El-Hajj, D. Mukherji, Y. N. Liu, G. Daoud and W. Abou-Kheir, Front. Oncol., 2018, 8, 347 CrossRef.
- J. Jin, B. Krishnamachary, Y. Mironchik, H. Kobayashi and Z. M. Bhujwalla, Sci. Rep., 2016, 6, 1–12 CrossRef.
- J. H. Kim, P. Verwilst, M. Won, J. Lee, J. L. Sessler, J. Han and J. S. Kim, J. Am. Chem. Soc., 2021, 143, 14115–14124 CrossRef CAS.
- L. Yang, P. Shi, G. Zhao, J. Xu, W. Peng, J. Zhang, G. Zhang, X. Wang, Z. Dong, F. Chen and H. Cui, Signal Transduction Targeted Ther., 2020, 5, 8 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available: 1H, 13C NMR and MS analyses of new compounds, experimental procedures, and supplementary figures. See DOI: 10.1039/d1cc05573a |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2022 |