
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Heterobimetallic uranyl(VI) alkoxides of lanthanoids: formation through simple ligand exchange†
Dennis
Grödler
 a,
Martin L.
Weidemann
b,
Andreas
Lichtenberg
a,
Tobias
Greven
a,
Robin
Nickstadt
a,
Malek
Haydo
a,
Mathias
Wickleder
a,
Axel
Klein
a,
Dirk
Johrendt
b,
Sanjay
Mathur
a,
Markus
Zegke
*a and
Aida
Raauf
*a
a,
Martin L.
Weidemann
b,
Andreas
Lichtenberg
a,
Tobias
Greven
a,
Robin
Nickstadt
a,
Malek
Haydo
a,
Mathias
Wickleder
a,
Axel
Klein
a,
Dirk
Johrendt
b,
Sanjay
Mathur
a,
Markus
Zegke
*a and
Aida
Raauf
*a
aDepartment of Chemistry, Institute for Inorganic Chemistry, University of Cologne, Greinstr. 6, 50939 Cologne, Germany. E-mail: m.zegke@uni-koeln.de; ajamil@uni-koeln.de
bDepartment of Chemistry, Ludwig-Maximilians-University of Munich, Butenandtstraße 5-13 (D), 81377 Munich, Germany
First published on 14th December 2021
Abstract
Lanthanoid and actinoid silylamides are versatile starting materials. Herein we show how a simple ligand exchange with tert-butanol leads to the formation of the first trimeric heterobimetallic uranyl(VI)–lanthanoid(III) alkoxide complexes. The μ3 coordination of the endogenous uranyl oxo atom results in a significant elongation of the bond length and a significant deviation from the linear uranyl arrangement.
Although the chemistry of high-valent uranyl (O
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) UO2+) accounts for 55% of all U structures reported in the CSD, not much attention has been paid to its alkoxide chemistry over the last 30 years. Only a few uranyl alkoxide compounds were structurally characterised in the 1950s by Bradley et al. and in the 1980s by Sattelberger et al.1,2 In contrast, far more alkoxide structures of the lower-valent uranium (U(IV)–U(V))3–6 species have been reported, because the preparation of uranyl alkoxides is accompanied by synthetic difficulties, e.g. solvolytic disproportionations, in which the cleavage of an oxo-unit takes place, while a second oxo-component is cleaved thermally to result in the formation of [U(OR)6] and [U2O5(OR)2(HOR)2].1 During the 1980s, coordinatively unsaturated alkoxides were reported to undergo ligand redistribution, resulting in oxo-alkoxide clusters.2 Mononuclear uranyl alkoxides could only be stabilised by coordinating sterically demanding ligands like Ph3PO by Burns et al. in 1992 and aryloxides by Barnhardt et al. in 1995.7,8 Even though uranyl-lanthanoid assemblies have been investigated in different systems,9–12 the heterobimetallic alkoxides have been rarely investigated, and, to the best of our knowledge, only few complexes such as [Li(THF)2U(OtBu)6], [Li(Et2O)U(OtBu)6] and [KU2(OtBu)9] have been reported, where alkali metals are bridged via OtBu ligands to U(IV) or U(V) centres and none of them contain uranyl (OUO2+) units.13,14 The compound [Zr2(OiPr)9U(C8H8)] prepared from [Zr2(OiPr)9]UI2(THF) by Evans in 2001 is the only known heterobimetallic alkoxide that contains U(III) and a transition metal.15 A CSD search on uranium bridged to lanthanoids via oxygen revealed only 19 examples, but none of these structures can be classified as an alkoxide. Most of them are reduced uranyl oxo-bridged complexes. In 2011 Arnold et al. first reported the reduction of a uranyl(VI) polypyrrolic Schiff-base macrocyclic complex [UO2(THF)(H2L)] by Sm(II) silylamide [SmN′′2(THF)2] (N′′ = N{Si(CH3)3}2) or by sterically induced reduction with YN′′3 resulting in [{UO2M(py)2(L)}2] (M = Y, Sm) (L = “Pacman” type calix pyrrole macrocycle). The series was later extended to M = Sc, Ce, Sm, Eu, Gd, Dy, Er, Yb and Lu in 2013.16,17 Additionally, they were also able to isolate a small series of linear, oxo-bridged trinuclear compounds e.g. [{UO2(py)5}2(LnI4)]I (Ln = Sm, Dy) containing a uranyl(V) by reduction of [UO2Cl2(THF)2] with Ln(II) (Ln = Sm, Dy) halides in 2017.18 The only other known compounds are phosphate- or mellitate-bridged clusters.19,20
UO2+) accounts for 55% of all U structures reported in the CSD, not much attention has been paid to its alkoxide chemistry over the last 30 years. Only a few uranyl alkoxide compounds were structurally characterised in the 1950s by Bradley et al. and in the 1980s by Sattelberger et al.1,2 In contrast, far more alkoxide structures of the lower-valent uranium (U(IV)–U(V))3–6 species have been reported, because the preparation of uranyl alkoxides is accompanied by synthetic difficulties, e.g. solvolytic disproportionations, in which the cleavage of an oxo-unit takes place, while a second oxo-component is cleaved thermally to result in the formation of [U(OR)6] and [U2O5(OR)2(HOR)2].1 During the 1980s, coordinatively unsaturated alkoxides were reported to undergo ligand redistribution, resulting in oxo-alkoxide clusters.2 Mononuclear uranyl alkoxides could only be stabilised by coordinating sterically demanding ligands like Ph3PO by Burns et al. in 1992 and aryloxides by Barnhardt et al. in 1995.7,8 Even though uranyl-lanthanoid assemblies have been investigated in different systems,9–12 the heterobimetallic alkoxides have been rarely investigated, and, to the best of our knowledge, only few complexes such as [Li(THF)2U(OtBu)6], [Li(Et2O)U(OtBu)6] and [KU2(OtBu)9] have been reported, where alkali metals are bridged via OtBu ligands to U(IV) or U(V) centres and none of them contain uranyl (OUO2+) units.13,14 The compound [Zr2(OiPr)9U(C8H8)] prepared from [Zr2(OiPr)9]UI2(THF) by Evans in 2001 is the only known heterobimetallic alkoxide that contains U(III) and a transition metal.15 A CSD search on uranium bridged to lanthanoids via oxygen revealed only 19 examples, but none of these structures can be classified as an alkoxide. Most of them are reduced uranyl oxo-bridged complexes. In 2011 Arnold et al. first reported the reduction of a uranyl(VI) polypyrrolic Schiff-base macrocyclic complex [UO2(THF)(H2L)] by Sm(II) silylamide [SmN′′2(THF)2] (N′′ = N{Si(CH3)3}2) or by sterically induced reduction with YN′′3 resulting in [{UO2M(py)2(L)}2] (M = Y, Sm) (L = “Pacman” type calix pyrrole macrocycle). The series was later extended to M = Sc, Ce, Sm, Eu, Gd, Dy, Er, Yb and Lu in 2013.16,17 Additionally, they were also able to isolate a small series of linear, oxo-bridged trinuclear compounds e.g. [{UO2(py)5}2(LnI4)]I (Ln = Sm, Dy) containing a uranyl(V) by reduction of [UO2Cl2(THF)2] with Ln(II) (Ln = Sm, Dy) halides in 2017.18 The only other known compounds are phosphate- or mellitate-bridged clusters.19,20
Particularly important in this context is also the activation of the uranyl(VI) dication, which, due it its OUO bond strength with a notional bond order of three and a mean U–O bond dissociation enthalpy of 604 kJ mol−1 is one of the most stable oxo-cations.21,22 Over the last 15 years the reduction chemistry of uranyl(VI) has seen a renaissance, and particularly the coordination of hetero metal atoms to the uranyl oxo atoms has resulted in a variety of reduced uranium complexes that show catalytic activity,23 cation-cation interactions (CCIs),24 thermally, photochemically or chemically reduced uranyl(V) centres and significantly elongated U–O bonds and paramagnetism attributable to the 5f1 state.25–28 Many of these complexes have been stabilised with auxiliary ligands, such as macrocycles, bipyridine/phenantroline-type29 or dipyrrin-type30 ligands but also with just coordinating solvent molecules.31
We have revisited this chemistry herein, and present the first heterobimetallic uranyl-lanthanoid alkoxides with the general formula [Ln2UO2(OtBu)n(L)] (L = OAc, n = 7; L = THF, n = 8), obtained by the reaction of [UO2(L)2(THF)2] (L = OAc, N′′) with the corresponding metal silylamides Ln(N′′)3 (Ln = Nd, Sm, Gd, Dy, Ho, Er, Yb) in THF followed by adding an excess of tert-butanol (Scheme 1). The products resulting from the reaction of [UO2(OAc)2(THF)2] with LnN′′3 are oligomers by acetate-bridging of the trinuclear-monomeric unit [Ln2UO2(OtBu)7(OAc)]x (x = 2–4) whereas the reaction of [UO2N′′2(THF)2] with LnN′′3 resulted in a merely trinuclear-monomeric complex.
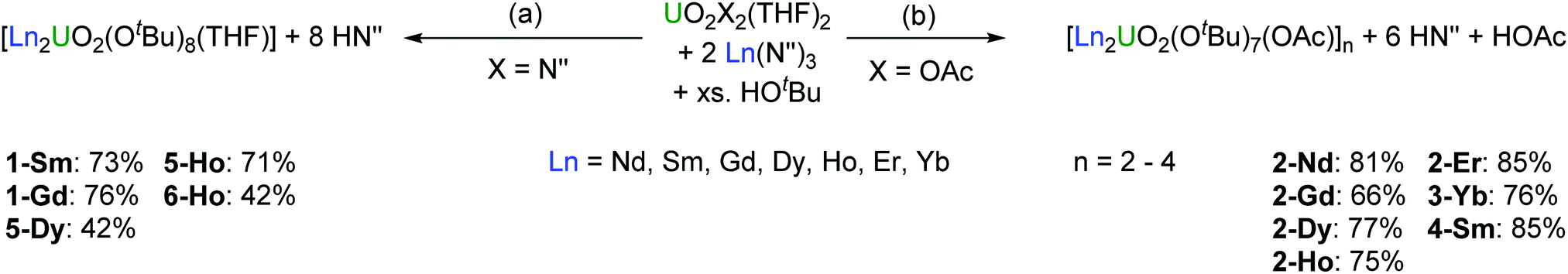 | ||
| Scheme 1 Synthesis of mixed uranyl-lanthanoid alkoxides by reaction of either [UO2N′′2(THF)2] or [UO2(OAc)2(THF)2] with the respective [LnN′′3] (Ln = Nd, Sm, Gd, Dy, Ho, Er, Yb) to afford the monomeric cluster [Ln2UO2(OtBu)8(THF)] (a) or the acetate-bridged [Ln2UO2(OtBu)7(OAc)]n (b). | ||
The compounds were analysed structurally by scXRD and were investigated by Physical Properties Measurement System (PPMS). We have been able to isolate a variety of clear red-orange single crystals by careful crystallisation from saturated benzene or toluene solutions at 4 °C.
All [Ln2UO2(OtBu)n(L)] (Ln = Nd, Gd, Dy, Ho; L = OAc, n = 7; L = THF, n = 8) complexes show similar bonding motifs (Fig. 1). The molecular structure consists of two lanthanoid centres and one uranyl subunit linked through three μ2-OtBu groups to form a triangle which is capped by one μ3-OtBu on the one side and by one uranyl oxygen (μ3-OendoUOexo) on the other side. Moreover, the uranyl centre possesses one terminal OtBu group to complete a distorted octahedral configuration and each Ln centre possesses one terminal OtBu-group. To saturate the coordination sphere of the Ln centre, the acetate group is bridged to the next [Ln2UO2(OtBu)7(OAc)] unit, resulting in the formation of an oligomeric structure [Ln2UO2(OtBu)7(OAc)]n (Scheme 1(b)). In contrast, starting from [UO2N′′2(THF)2] results, due to the absence of the acetate group, in a Ln atom possessing an additional terminal OtBu group. The coordination sphere of the second Ln centre is completed by a THF molecule resulting in the trinuclear complex [Ln2UO2(OtBu)8(THF)] (Scheme 1(a)) displaying a distorted octahedral geometry around each Ln. In addition, the endogenous uranyl Oμ3UO bond lengths are elongated to 1.866–1.902 Å, respectively, when compared to non-oxo-coordinated uranyl compounds (OUO 1.78 Å) (see Fig. S29, ESI†). This is in the range of various uranyl(V) complexes,32 even though C. J. Burns et al. have shown that significant uranyl lengthening and bending can occur in uranyl(VI) oxometallate clusters.33 We can only hypothesise at this stage that this is due to the smaller radii of later lanthanoid ions, which are harder Lewis acids and therefore a better acceptor for the also hard uranyl oxygen. The OUO bond angle is also bent to below 170°. Previous studies have shown that the OUO unit can deviate significantly from a linear arrangement given the right ligand environment. For further details see the overview by Hayton and the references therein.34 The latter study also shows that this influences the colour of the compound. As deviations from the linear arrangement are rare it is quite remarkable that our samples consistently show OUO bond angles below 170° (see Fig. S30, ESI†).
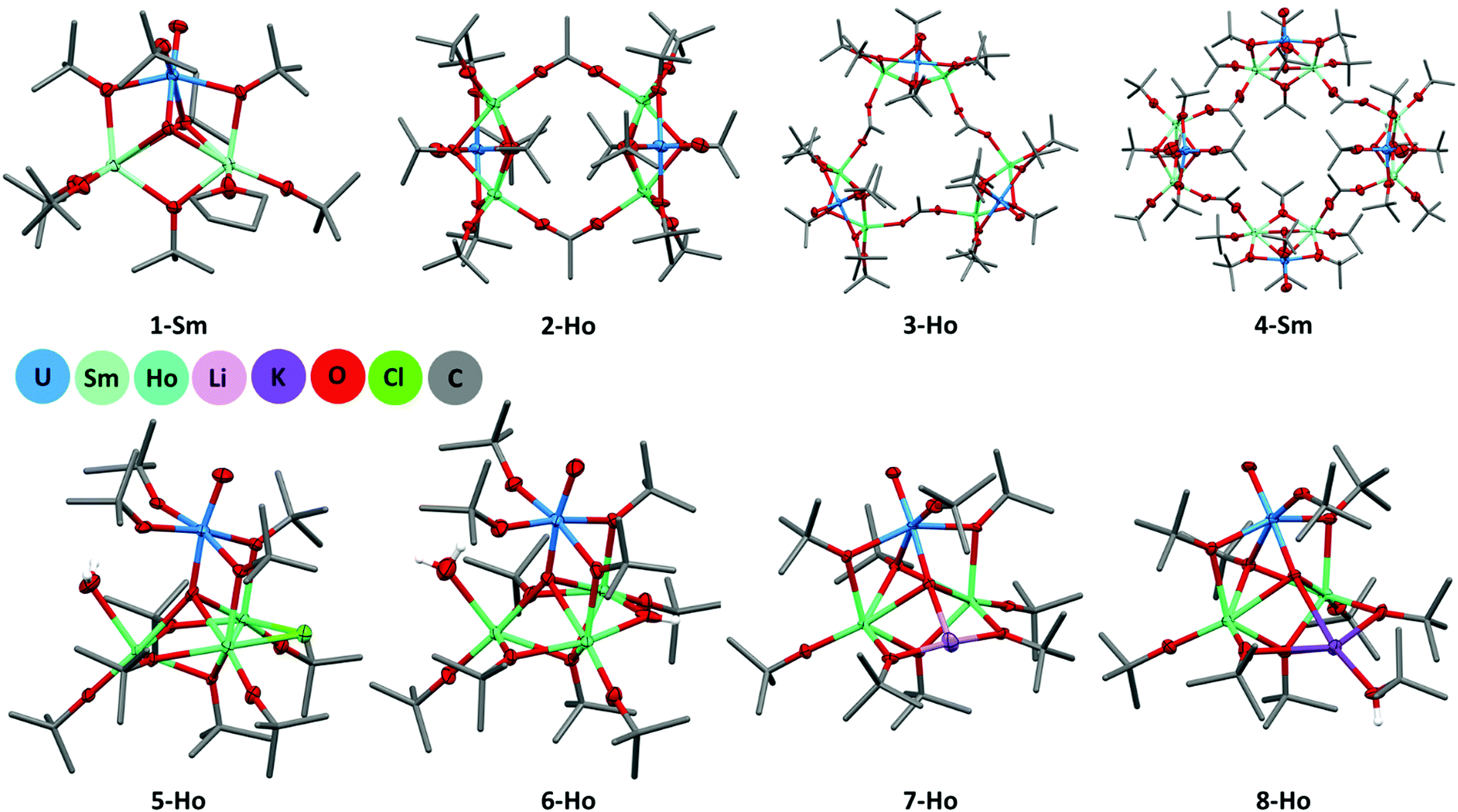 | ||
| Fig. 1 Selected molecular structure of the monomeric 1-Sm, dimeric 2-Ho, trimeric 3-Ho, tetrameric 4-Sm, chloride and hydroxide inclusion 5-Ho and 6-Ho and alkali metal inclusion 7-Ho and 8-Ho alkoxides. Thermal ellipsoids are shown at 50% probability level and hydrogen atoms have been omitted for clarity. | ||
The trinuclear “Ln2U” unit resembles the molecular structure of previously reported trimeric Ln tert-butoxides [Ln3(OtBu)9(THF)2]35 with one Ln component being replaced by the uranyl unit in our compounds, and consequently a tridentate OtBu group and a coordinating THF molecule are replaced by the uranyl oxygen units (Scheme 2).
 | ||
| Scheme 2 Illustrations of the molecular structure of (a) trinuclear monomeric unit [Ln2UO2(OtBu)7(OAc)]n, (b) trinuclear compound [Ln2UO2(OtBu)7(THF)] and (c) comparison with the homometallic lanthanoid tert-butoxide [Ln3(OtBu)9(THF)2]. | ||
Comparing the Ln series we observed that with increasing atomic number the Ln–Oμ3 coordination shortens from 2.626 Å (2-Nd) to 2.447 Å (3-Yb) as a consequence of the decreasing ionic radius. This results in an elongation of Oμ3U to about 1.88 Å in 3-Yb (see Table S1, ESI†). In addition, we found different crystals of the material leading to an expanded connectivity between the acetate-bridges, depending on how long the complexes were crystallised for. When the compound was crystallised in a period of 1–2 days only crystals of monomeric constitution were isolated, but after two weeks clusters with up to four [Ln2UO2(OtBu)7(OAc)] units were found for Ln = Sm and three [Ln2UO2(OtBu)7(OAc)] units for Ln = Ho and Yb, the latter crystallising in the cubic space group Pa![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) (see SI for details).
(see SI for details).
In addition, the reaction between [UO2N′′2(THF)2] and LnN′′3 is also prone to side reactions if unsublimed starting materials are used, as observed by the isolation of complexes with chloride or alkali metal inclusions (Fig. 1). Because these complexes contain unique connectivity of the metal centres, these were then made on purpose. To provide 1 equiv. of chloride 1/3 equiv. of the respective LnCl3·6H2O salt was added to a reaction mixture with sublimed LnN′′3. Utilising the hydrated LnCl3 salt proved to be a useful method to keep the content of water as low as possible but at the same time providing a coordinating water molecule which is a necessary feature of [Dy3UO2(OtBu)10Cl(H2O)] 5-Dy and [Ho3UO2(OtBu)10Cl(H2O)] 5-Ho. Both 5-Dy and 5-Ho could not be isolated when water was completely excluded even though THF is present in the reaction mixture that could saturate the coordination sphere of the Ln centre. By looking at the space filling of the tert-butanol ligands it becomes evident that a THF molecule does not have enough space in this gap. 5-Dy and 5-Ho both crystallise in the orthorhombic space group Pnma. Their structures can be described as an [UO2(OtBu)2] moiety binding to a [Ln3(OtBu)8Cl] trimer via coordination of the uranyl oxygen to three lanthanoid centres and two bridging μ2-OtBu to two of the three Ln atoms. The third Ln atom shows a coordinating water molecule to saturate the coordination sphere. The bond distances of 2.481(4) Å for both 5-Dy and 5-Ho fit well with other Ln water bonds found in the literature and are in accordance with the oxidation state of the metal centres.36,37 Alkali metal inclusions were reproduced by adding one additional equiv. of MN′′ (M = Li, K) to a reaction mixture of [UO2N′′2(THF)2] and LnN′′3. Because of the small ionic radius of Li, it fits into the small gap of the former endogenous tert-butoxides, whereas K possesses a protonated tert-butanol ligand to saturate the coordination sphere. In general, we found that complexes containing later Ln decompose rather quickly, sometimes before crystallisation can set in, identified by a colour change from orange to red, or undergo side reactions more readily than those with early Ln. The UV-vis absorption spectra (see Fig. S26, ESI†) for [Gd2UO2(OtBu)7(OAc)]2 (2-Gd) and [Ho2UO2(OtBu)7(OAc)]2 (2-Ho) showed an absorption at 270 nm for (2-Gd) and 275 nm for 2-Ho, respectively. Additional bands were observed at 362 nm and 450 nm. The pattern of the UV-vis absorption spectra is in accordance with the spectra of uranyl formohydroxamate reported by Albrecht-Schmitt et al. but show a hypsochromic shift to 450 nm corresponding to a low intensity charge transfer band compared to typical uranyl compounds.21,38 The hydrolysis curve for 2-Ho was determined by the decreasing absorption of the complex after exposure to atmospheric moisture (see Fig. S27 and S28, ESI†).
PPMS studies of the susceptibility and magnetisation between 2.1 and 300 K confirmed that [Gd2UO2(OtBu)7(OAc)]2 (2-Gd), [Dy2UO2(OtBu)7(OAc)]2 (2-Dy) and [Ho2UO2(OtBu)7(OAc)]2 (2-Ho) are paramagnetic according to the Curie–Weiss rule39 with effective magnetic moments of 7.92(1), 10.36(1) and 10.32(1) μB, respectively. These values agree with the expected moments for Gd3+, Dy3+ and Ho3+ (Table 1), which suggests, considering charge neutrality with uranium being U(VI), that the rare-earth ions are the only active magnetic species in these compounds.
| Magnetic centres | μ expeff (μB) | n expeff | g J [J(J + 1)]1/2 |
|---|---|---|---|
| Gd3+ | 7.92(1) | 7.8–7.9 | 7.94 |
| Dy3+ | 10.36(1) | 10.2–10.6 | 10.65 |
| Ho3+ | 10.32(1) | 10.3–10.5 | 10.61 |
Negative values of the Weiss constant θ (Fig. S24, ESI†) and a slight flattening of χmol below 4 K indicate antiferromagnetic ordering of the Dy and Ho compounds.41 In case of the Gd containing complex a negative θ value suggests antiferromagnetic ordering as well, though the transition temperature is apparently below the lower measurement limit.
Isothermal magnetisation plots (Fig. S25, ESI†) are linear with the field at 300 K. In case of [Gd2UO2(OtBu)7(OAc)]2 the curve saturates at a value near gJJ = 7 for Gd3+. The magnetisation of the Dy and Ho samples saturate against 5 μB, which is only half of the expected value gJJ = 10 for Dy3+ and Ho3+, possibly due to the antiferromagnetic order.
In conclusion, we have isolated the first trimeric heterobimetallic uranyl(VI)–lanthanoid(III) alkoxide complexes via a simple and straightforward ligand exchange. The μ3 coordination of the endogenous uranyl oxo atom results in a significant elongation of the bond length and a significant deviation from the linear uranyl arrangement. This indicates that a coordination of lanthanoid atoms to the uranyl oxo-atoms may facilitate the reduction chemistry towards lower oxidation states of uranium, resulting in trimetallic systems with 5f–4f electron correlations. The magnetic susceptibilities obey the Curie–Weiss rule with effective magnetic moments compatible to Gd3+, Dy3+ and Ho3+, respectively, and indicate antiferromagnetic ordering below 4 K for the Dy3+ and Ho3+ compounds.
D. G.: Original draft, data curation, formal analysis, investigation, visualisation; M. W., A. L., T. G., R. N., M. H.: Data curation, formal analysis, investigation; S. M., M. W., A. K., D. J.: Funding acquisition, supervision, validation, resources, review & editing; M. Z., A. R.: conceptualisation, methodology, project administration, review & editing, supervision.
The authors thank Mario Winkler, Jonathan Wischnat and Prof. Dr Joris van Slageren, University of Stuttgart, for help with physicochemical measurements. The authors thanks Mr Dirk Pullem for carrying out the elemental analyses.
Conflicts of interest
There are no conflicts of interest to declare.Notes and references
- D. C. Bradley, A. K. Chatterjee and A. K. Chatterjee, J. Inorg. Nucl. Chem., 1959, 12, 71–78 CrossRef CAS.
- C. J. Burns and A. P. Sattelberger, Inorg. Chem., 1988, 27, 3692–3693 CrossRef CAS.
- R. G. Jones, G. Karmas, G. A. Martin and H. Gilman, J. Am. Chem. Soc., 1956, 78, 4285–4286 CrossRef CAS.
- P. G. Eller and P. J. Vergamini, Inorg. Chem., 1983, 22, 3184–3189 CrossRef CAS.
- F. A. Cotton, D. O. Marler and W. Schwotzer, Inorg. Chim. Acta, 1984, 95, 207–209 CrossRef CAS.
- F. A. Cotton, D. O. Marler and W. Schwotzer, Inorg. Chim. Acta, 1984, 85, L31–L32 CrossRef CAS.
- C. J. Burns, D. C. Smith, A. P. Sattelberger and H. B. Gray, Inorg. Chem., 1992, 31, 3724–3727 CrossRef CAS.
- D. M. Barnhart, C. J. Burns, N. N. Sauer and J. G. Watkin, Inorg. Chem., 1995, 34, 4079–4084 CrossRef CAS.
- P. Thuéry, Inorg. Chem., 2009, 48, 825–827 CrossRef PubMed.
- J. A. Ridenour and C. L. Cahill, CrystEngComm, 2018, 20, 4997–5011 RSC.
- P. Thuéry, CrystEngComm, 2012, 14, 3363–3366 RSC.
- P. Thuéry, CrystEngComm, 2008, 10, 1126–1128 RSC.
- S. Fortier, G. Wu and T. W. Hayton, Inorg. Chem., 2008, 47, 4752–4761 CrossRef CAS PubMed.
- W. G. Van der Sluys, A. P. Sattelberger and M. W. McElfresh, Polyhedron, 1990, 9, 1843–1848 CrossRef CAS.
- W. J. Evans, G. W. Nyce, M. A. Greci and J. W. Ziller, Inorg. Chem., 2001, 40, 6725–6730 CrossRef CAS PubMed.
- P. L. Arnold, E. Hollis, F. J. White, N. Magnani, R. Caciuffo and J. B. Love, Angew. Chem., Int. Ed., 2011, 50, 887–890 CrossRef CAS PubMed.
- P. L. Arnold, E. Hollis, G. S. Nichol, J. B. Love, J.-C. Griveau, R. Caciuffo, N. Magnani, L. Maron, L. Castro, A. Yahia, S. O. Odoh and G. Schreckenbach, J. Am. Chem. Soc., 2013, 135, 3841–3854 CrossRef CAS PubMed.
- P. L. Arnold, B. E. Cowie, M. Suvova, M. Zegke, N. Magnani, E. Colineau, J.-C. Griveau, R. Caciuffo and J. B. Love, Angew. Chem., Int. Ed., 2017, 56, 10775–10779 CrossRef CAS PubMed.
- K. E. Knope, D. T. de Lill, C. E. Rowland, P. M. Cantos, A. de Bettencourt-Dias and C. L. Cahill, Inorg. Chem., 2012, 51, 201–206 CrossRef CAS PubMed.
- C. Volkringer, N. Henry, S. Grandjean and T. Loiseau, J. Am. Chem. Soc., 2012, 134, 1275–1283 CrossRef CAS PubMed.
- R. G. Denning, J. Phys. Chem. A, 2007, 111, 4125–4143 CrossRef CAS PubMed.
- J. K. Gibson, R. G. Haire, M. Santos, J. Marçalo and A. Pires de Matos, J. Phys. Chem. A, 2005, 109, 2768–2781 CrossRef CAS PubMed.
- N. Behera and S. Sethi, Eur. J. Inorg. Chem., 2021, 95–111 CrossRef CAS.
- V. Mougel, L. Chatelain, J. Pécaut, R. Caciuffo, E. Colineau, J.-C. Griveau and M. Mazzanti, Nat. Chem., 2012, 4, 1011–1017 CrossRef CAS PubMed.
- M. Zegke, G. S. Nichol, P. L. Arnold and J. B. Love, Chem. Commun., 2015, 51, 5876–5879 RSC.
- P. L. Arnold, M. S. Dutkiewicz, M. Zegke, O. Walter, C. Apostolidis, E. Hollis, A.-F. Pécharman, N. Magnani, J.-C. Griveau, E. Colineau, R. Caciuffo, X. Zhang, G. Schreckenbach and J. B. Love, Angew. Chem., Int. Ed., 2016, 55, 12797–12801 CrossRef CAS PubMed.
- M. Zegke, X. Zhang, I. Pidchenko, J. A. Hlina, R. M. Lord, J. Purkis, G. S. Nichol, N. Magnani, G. Schreckenbach, T. Vitova, J. B. Love and P. L. Arnold, Chem. Sci., 2019, 10, 9740–9751 RSC.
- B. E. Cowie, J. M. Purkis, J. Austin, J. B. Love and P. L. Arnold, Chem. Rev., 2019, 119, 10595–10637 CrossRef CAS PubMed.
- P. Thuéry and J. Harrowfield, Dalton Trans., 2017, 46, 13660–13667 RSC.
- J. R. Pankhurst, N. L. Bell, M. Zegke, L. N. Platts, C. A. Lamfsus, L. Maron, L. S. Natrajan, S. Sproules, P. L. Arnold and J. B. Love, Chem. Sci., 2017, 8, 108–116 RSC.
- L. Natrajan, F. Burdet, J. Pécaut and M. Mazzanti, J. Am. Chem. Soc., 2006, 128, 7152–7153 CrossRef CAS PubMed.
- P. L. Arnold, J. B. Love and D. Patel, Coord. Chem. Rev., 2009, 253, 1973–1978 CrossRef CAS.
- P. B. Duval, C. J. Burns, D. L. Clark, D. E. Morris, B. L. Scott, J. D. Thompson, E. L. Werkema, L. Jia and R. A. Andersen, Angew. Chem., Int. Ed., 2001, 40, 3357–3361 CrossRef CAS PubMed.
- T. W. Hayton, Dalton Trans., 2018, 47, 1003–1009 RSC.
- J. Gromada, A. Mortreux, T. Chenal, J. W. Ziller, F. Leising and J.-F. Carpentier, Chem. – Eur. J., 2002, 8, 3773–3788 CrossRef CAS.
- T. J. Boyle, M. L. Neville, J. M. Sears, R. E. Cramer, M. A. Rodriguez, T. M. Alam and S. P. Bingham, Polyhedron, 2016, 118, 52–60 CrossRef CAS.
- Y. Jiang, G. Brunet, R. J. Holmberg, F. Habib, I. Korobkov and M. Murugesu, Dalton Trans., 2016, 45, 16709–16715 RSC.
- M. A. Silver, W. L. Dorfner, S. K. Cary, J. N. Cross, J. Lin, E. J. Schelter and T. E. Albrecht-Schmitt, Inorg. Chem., 2015, 54, 5280–5284 CrossRef CAS PubMed.
- A. S. Arrott, Phys. Rev. B: Condens. Matter Mater. Phys., 1985, 31, 2851–2856 CrossRef CAS PubMed.
- H. Lueken, Magnetochemie, Vieweg + Teubner Verlag, 1st edn, 1999 Search PubMed.
- P. W. Anderson, Phys. Rev., 1950, 79, 705–710 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2110472–2110482, 2111075, 2111783–2111784. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1cc05444a |
| This journal is © The Royal Society of Chemistry 2022 |