
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Rubidium and caesium aluminyls: synthesis, structures and reactivity in C–H bond activation of benzene†
Thomas X.
Gentner
a,
Matthew J.
Evans
b,
Alan R.
Kennedy
 a,
Sam E.
Neale
c,
Claire L.
McMullin
*c,
Martyn P.
Coles
*b and
Robert E.
Mulvey
*a
a,
Sam E.
Neale
c,
Claire L.
McMullin
*c,
Martyn P.
Coles
*b and
Robert E.
Mulvey
*a
aDepartment of Pure and Applied Chemistry, University of Strathclyde, Glasgow, G1 1XL, UK. E-mail: r.e.mulvey@strath.ac.uk
bSchool of Chemical and Physical Sciences, Victoria University of Wellington, P.O. Box 600, Wellington, New Zealand. E-mail: martyn.coles@vuw.ac.nz
cDepartment of Chemistry, University of Bath, Bath, BA2 7AY, UK. E-mail: c.mcmullin@bath.ac.uk
First published on 7th January 2022
Abstract
Expanding knowledge of low valent aluminium chemistry, rubidium and caesium aluminyls are reported to complete the group 1 (Li–Cs) set of metal aluminyls. Both compounds crystallize as a contacted dimeric pair supported by M⋯π(arene) interactions with a pronounced twist between aluminyl units. Density functional theory calculations show symmetrical bonding between the M and Al atoms, with an Al centred lone-pair donating into vacant Rb and Cs orbitals. Interestingly, despite their structural similarity the Cs aluminyl enables C–H bond activation of benzene, but not the Rb aluminyl reflecting the importance of the alkali metal in these heterobimetallic systems.
A recent review highlighted the growing recognition of alkali metal mediation, AMM, that underpins a diversity of applications in main group organometallic chemistry. AMM can be defined as chemical transformations that cannot occur at all or cannot take place efficiently, without intervention of an alkali metal.1 Often AMM is manifest in bimetallic formulations that contain an alkali metal partnered by a second metal, the unique properties of which stem from cooperative effects between the two metals.2,3 One emerging area that is benefitting from AMM is low valent aluminium chemistry, specifically the new class of aluminyl anions, [Al{L}n]− ({L}n = bidentate, dianionic ancillary ligand framework).4 Access to these highly reactive compounds is principally achieved via reduction of a suitably supported aluminium(III) precursor using potassium metal5,6 or KC8,7 while syntheses from Al(II)8–10 and Al(I)11 starting materials are also known. In most cases the potassium plays a dual role in this process, acting as both an effective reagent for the reduction of Al(III) to Al(I) and as an integral stabilising component of the potassium aluminyl product, K[Al{L}n].
Aluminyls may be classified according to their structure in a manner that is correlated with the nature and extent of M···Al interactions (M = Li, Na, K). This is illustrated for the [Al(NONDipp)]− system, where three distinct structural types are known for the potassium salts (Fig. 1).12 Reduction of the aluminium(III) iodide Al(NONDipp)I with potassium metal affords the donor-solvent free contacted dimeric pair (CDP) I.5 This structure type is common for aluminyls in which aryl-substituents are present that allow K⋯π(arene) interactions.5–7,11 Recent work has shown that the dimeric structure of I can be cleaved using TMEDA to afford a monomeric ionic pair (MIP) II, a structure related to that observed for a dialkyl-substituted aluminyl.8 Furthermore, encapsulation of the potassium cation in I using [2.2.2]cryptand affords the separated ion pair (SIP) III,10,12 representing a rare example of this motif in aluminyl chemistry where no interaction exists between aluminium and potassium.9,10,13
 | ||
| Fig. 1 A series of potassium aluminyls illustrating the three distinct major structural forms of CDP, MIP and SIP. (2.2.2)crypt = [2.2.2]cryptand. | ||
Far from being superficial, these distinct structural types can impart profound differences on the chemical reactivity of aluminyls, demonstrating the importance of AMM in this area. This is best illustrated with the contrasting reactivity of the xanthene supported system [Al(XanthNONDipp)]− (XanthNONDipp = [4,5-(NDipp)2-2,7-tBu2-9,9-Me2-xanthene]2−) with benzene. It was shown that the potassium CDP thermally activated a C–H bond,7 whereas the corresponding [K(2.2.2)crypt]+ SIP reversibly cleaved a C–C bond of the aromatic ring to afford the seven-membered AlC6H6 metallacycle.13
We have recently extended this field to show that the [Al(NONDipp)]− aluminyl anion can be accessed directly from reduction of Al(III) iodide using lithium and sodium metal.14 The products isolated from non-coordinating solvent exist as ‘slipped’ CDPs (Scheme 1, IV-Li and IV-Na), with DFT studies confirming only one bond from aluminium to the alkali metal. Furthermore, we demonstrated that addition of Et2O cleaved the CDP to afford the corresponding solvated MIPs V-Li and V-Na, containing unsupported Al–Li and Al–Na bonds.15,16 In the knowledge that systematic studies spanning the whole of group 1 (Li–Cs) are still relatively rare,17 and that often substitution of one alkali metal for another can have a profound effect on structure and reactivity, in this contribution we complete the series of alkali metal aluminyls (Li–Cs), with the first report of rubidium and caesium aluminyls, [M{Al(NONDipp)}]2.
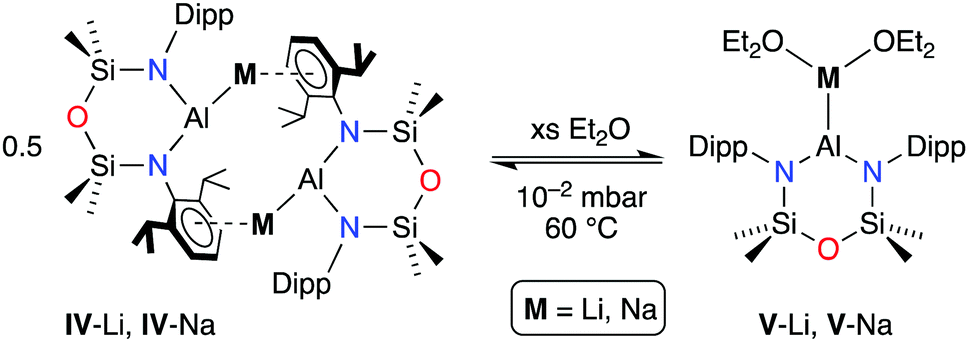 | ||
| Scheme 1 The solution-state equilibrium between slipped CDPs (IV) and MIPs (V) for lithium and sodium aluminyls. | ||
To access the rubidium and caesium aluminyls, Al(NONDipp)I was reduced in C6H6 with RbC8 and CsC8, respectively (Scheme 2). In each case, the initially colourless solution turned yellow after stirring at room temperature overnight. The reaction proceeded smoothly and the 1H NMR spectra of the products from Rb (1) and Cs (2) reductions compared well to that of their lighter congeners [M{Al(NONDipp)}]2 (M = Li, Na, K),5,14 showing a high-field singlet for the SiMe2 groups (0.36 ppm) consistent with C2h-symmetry (Fig. S1 and S3, ESI†). As for all other aluminyl systems reported to date, no signals were observed in the 27Al NMR spectra. The diffusion coefficients D of 1 (4.35 × 10−10 m2 s−1) and 2 (5.60 × 10−10 m2 s−1) obtained by 1H diffusion-ordered NMR spectroscopy (DOSY, C6D6, 294 K) are in the same range as those of the Li (5.32 × 10−10 m2 s−1), Na (4.60 × 10−10 m2 s−1) and K (4.48 × 10−10 m2 s−1) congeners. Both values are lower than that of the monomeric Al(III) iodide I (6.14 × 10−10 m2 s−1) indicating that the CPD structure is retained in aromatic solvents.
 | ||
| Scheme 2 Synthesis of the rubidium and caesium aluminyls, 1 and 2. | ||
Crystals suitable for X-ray crystallography were obtained by slowly cooling a saturated hexane solution from 60 °C to 5 °C overnight (1, M = Rb; 2, M = Cs). Both compounds crystallize as the centrosymmetric CDP (Fig. 2 and Table 1) and are isostructural with the congeneric potassium aluminyl [K{Al(NONDipp)}]2. Interestingly, the structures are isomorphous with the potassium indyl [K{In(NONDipp)}]2,18 suggesting that it is the sum of the ionic radii that influences the crystallographic symmetry, not the size of the individual metals (ΣK⋯In 3.45 Å, cf. ΣK⋯Al 3.24 Å, ΣRb⋯Al 3.41 Å, ΣCs⋯Al 3.65 Å).19 Consistent with the aluminyl salts of the smaller alkali metals, there is no interaction between the aluminium and the oxygen atom (mean Al⋯O: 1, 3.418 Å; 2, 3.431 Å) and the NONDipp ligand is strictly κ2-N,N′ with mean Al–N bond distances of 1.894 Å (1) and 1.898 Å (2) (Li: 1.875 Å; Na: 1.874 Å; K: 1.885 Å). The two anionic [Al(NONDipp)]− units are linked by Rb+/Cs+⋯π-(arene) interactions, with mean M⋯Ct (Ct = centroid) distances of 3.120 Å and 3.257 Å, respectively (Rb⋯C range 3.339(2)–3.472(2) Å, av. 3.42 Å; Cs⋯C range 3.505(3)–3.594(3) Å, av. 3.55 Å). These values are comparable with those for structures in which Rb and Cs cations are located in similar binding pockets defined by flanking arene groups.20–22
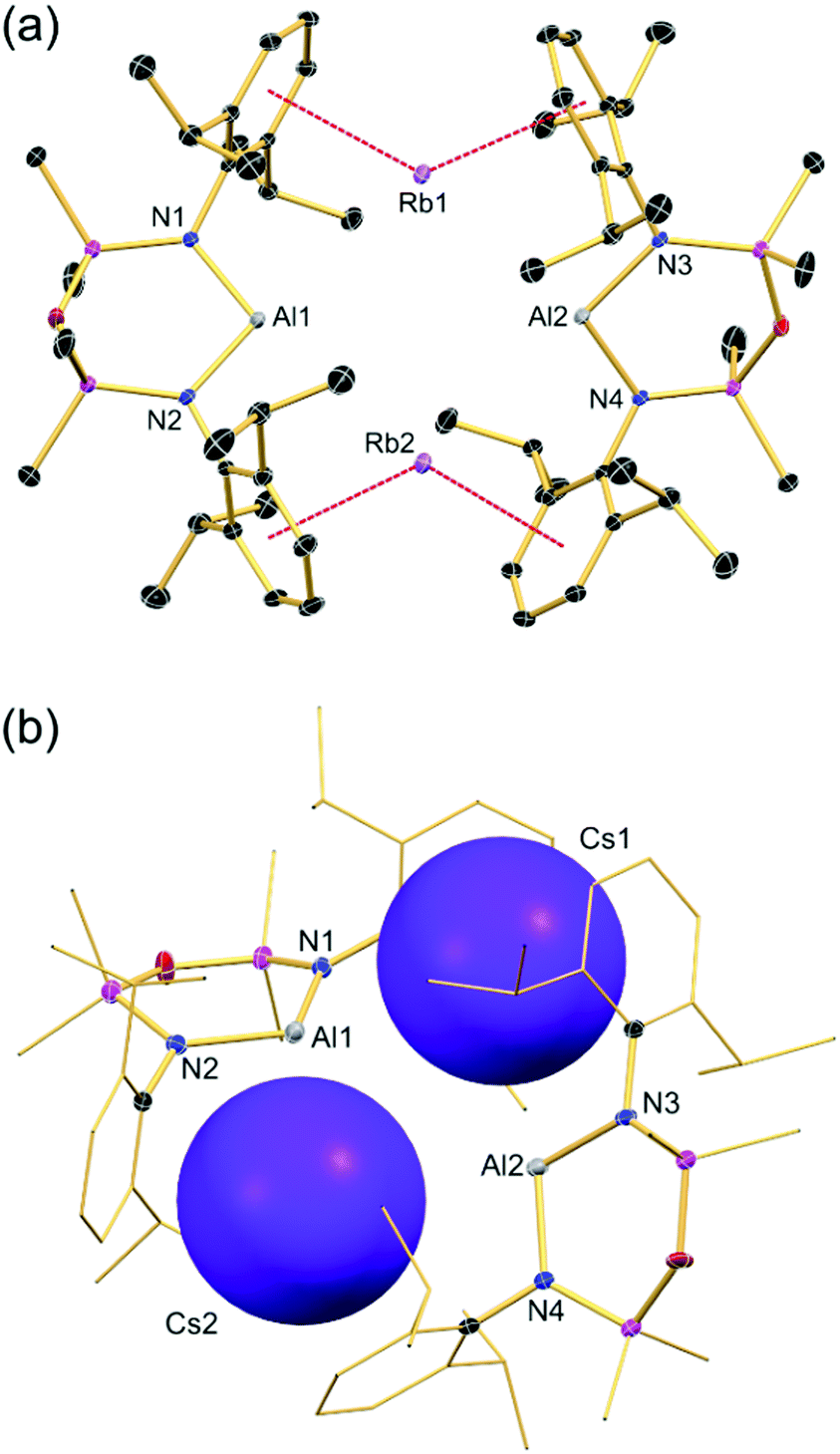 | ||
| Fig. 2 Thermal displacement plot (30% probability, H-atoms and hexane solvate omitted) of (a) [Rb{Al(NONDipp)}]21 and (b) [Cs{Al(NONDipp)}]2 (2, C-atoms represented as sticks and Cs atoms represented as space fill models to emphasize the twist angle θ). | ||
| Li | Na | K | Rb (1) | Cs (2) | |
|---|---|---|---|---|---|
| a Ct = centroid defined by six-membered aryl ring of Dipp-substituent. b Twist angle defined by the Al–N–Si–O–Si–N means square plane of each Al(NONDipp) group. | |||||
| Al⋯M | 2.746(3) | 3.0305(6) | 3.5346(8) | 3.7069(6) | 3.8833(10) |
| 3.364(3) | 3.5606(6) | 3.6437(9) | 3.7143(6) | 3.8960(9) | |
| 3.5916(8) | 3.7430(7) | 3.9001(10) | |||
| 3.7053(9) | 3.7678(7) | 3.9145(10) | |||
| Al⋯Al | 5.298(1) | 5.7097(6) | 5.673(1) | 5.548(1) | 5.752(1) |
| M⋯M | 3.108(8) | 3.3352(13) | 4.4950(8) | 4.9127(3) | 5.1406(3) |
| M⋯Cta | 2.008(4) | 2.4596(8) | 2.9004(10) | 3.0966(9) | 3.2474(14) |
| 3.0226(11) | 3.111(9) | 3.2505(14) | |||
| 3.030(6) | 3.1278(10) | 3.2601(14) | |||
| 3.0666(11) | 3.1428(10) | 3.2688(15) | |||
| θ | 0 | 0 | 33.55(4) | 66.55(4) | 66.31(5) |
To deal with the increasing size of the cation whilst still maintaining the beneficial, stabilizing M⋯π(arene) interactions, the Al(NONDipp) units are twisted away from each other, as shown by a larger angle (θ) between the Al–N–Si–O–Si–N planes (1:66.55(4)°; 2:66.31(5)°) when compared with the potassium congener (θ = 33.55(4)°). The much increased θ values in 1 and 2 enable the accommodation of the larger M+ cations between the Al(NONDipp) fragments without imposing a significant change to the Al⋯Al distances, which show little variation (M = K, 5.673(1) Å; M = Rb, 5.548(1) Å; M = Cs, 5.752(1) Å). This twisting also enables short contacts between the Al and the corresponding alkali metal, with mean values of 3.733 Å for Rb and for Cs 3.899 Å, which are both slightly larger than the sum of covalent radii (Σcov(Al, Rb) = 3.41; Σcov(Al, Cs) = 3.65).19
Next we examined the nature of the bonding in 1 and 2 using DFT calculations and compared the results with known CDPs M2[Al(NONDipp)]2 (M = Li, Na, K). The bond parameters of the optimized structures were in good agreement with the X-ray diffraction data, with calculated twist angles θ of 67.52° and 69.69° for the Rb and Cs CDPs, respectively. QTAIM analysis identified bond critical points (BCPs) (ρ(r) = 0.0081) between each Al and the alkali metal centres (Fig. 3), confirming the presence of four Al⋯M bonding interactions in the twisted CDPs. This is consistent with [K{Al(NONDipp)}]2 and in contrast to M = Li and Na for which only one Al···M BCP, albeit with higher covalent character, was present per Al. These bonding interactions in 1 and 2 are also implied from the Al–M Wiberg bond indices of 0.1327 (1) and 0.1430 (2) (Table S7, ESI†). As expected, and noted for the other members of the series,14 the M···Al bonding is essentially non-covalent in both CDPs,23,24 with a lower degree of covalency for the heavier alkali metals (−G(r)/V(r) ratios: Cs 1.06, Rb 1.08, K 1.06, Na 0.98, Li 0.88).25 The Laplacian of the electron densities for the Al2M2 core of 1 and 2 indicate areas of charge accumulation at aluminium, suggesting the presence of lone-pairs of electrons located at each Al centre. Indeed, NBO analysis identifies two NBOs which possess dominant lone-pair character on each Al centre in 1 and 2, each with significantly more s-orbital character over p-orbital character (1:83.1% s, 16.9% p; 2:84.1% s, 15.9% p). These interact with vacant Rb and Cs NBOs in 1 and 2, respectively, where second-order perturbation energy analysis identifies moderate donor–acceptor interactions (ΔE(2)Rb–Al[1] ≈ 10.2 kcal mol−1, ΔE(2)Cs–Al[2] ≈ 8.3 kcal mol−1). The natural atomic charges of the alkali metal atoms generally increase as the group is descended (Tables S3 and S4, ESI†), reflecting the lower electronegativity of the larger elements.
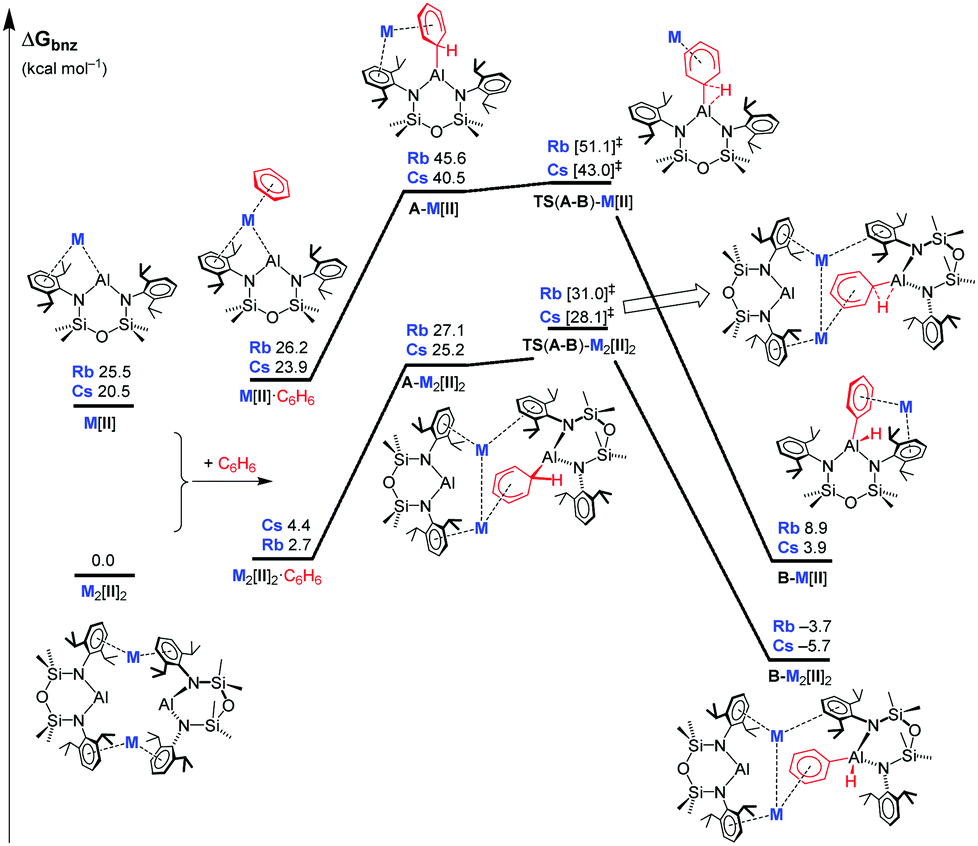 | ||
| Fig. 3 DFT-calculated free energy profile (BP86-D3BJ/BS2(C6H6)//BP86/BS1) in kcal mol−1 for the C–H activation of benzene relative to 1 (M = Rb) or 2 (M = Cs). See Fig. S25 (ESI†) for a larger version. | ||
AMM is clearly present in the reaction between H2 and [M{Al(NONDipp)}]2 (M = Li, Na, K) since hydrogenation (100 °C, 1.5 bar) proceeded in the order Li (t1/2 = 1.5 days) ≫ Na (t1/2 = 6 days) > K (t1/2 = 12 days). The hard lithium seems to be a perfect activator for H2, whereas descending the group the alkali metals become softer and thus reaction times increase. To check if AMM is present in 1 and 2, we investigated their reactivity towards benzene. While heating a C6H6 solution of 1 at 80 °C for several days remained unchanged, switching to 2 gave solid 3 after 5 days. The 1H NMR spectrum of 3 is consistent with forming [Cs{Al(NONDipp)(H)(Ph)}]nvia oxidative cleavage of a C–H bond of benzene, as indicated by new signals in the aromatic region reminiscent of a Ph ligand. The signal for the new hydride ligand could not be observed in the 1H NMR spectrum of 3.26 However an IR spectrum showed an Al–H stretch at 1685 cm−1 indicative of a hydride ligand attached to Al in aluminyl systems.7,26 Repeating the reaction in C6D6 gave further proof of bond activation since the C6H5 resonances for the phenyl ligand are no longer observed in the 1H NMR spectrum, and the Al–H stretch is replaced by an Al–D stretch that is shifted into the fingerprint region of the IR spectrum. Also the 1H coupled 27Al NMR spectrum shows a full width half-maximum of 506 Hz, 125 Hz greater than in the 27Al{1H} and is attributed to unresolved 1JAlH coupling (Fig. S10, ESI†).
Although aromatic C–H bond activation has been observed in aluminyl systems,7,8,11,27,282 is to date the only compound in the series of [M{Al(NONDipp)}]2 (M = Li–Cs) that enables this reactivity pointing towards a significant synergistic effect. A preliminary DFT study carried out on the C–H activation of benzene by 1 and 2 support these findings (Fig. 3). In both cases, formation of the constituent monomers M[II] from the CDPs is endergonic (Rb, 25.5 kcal mol−1; Cs, 20.5 kcal mol−1) and although solvation with benzene stabilizes the hypothetical ‘M[Al(NONDipp)]·n(C6H6)’ monomer, these products computed for Cs are higher in free energy by 23.9, 11.2 and 13.0 kcal mol−1 for n = 1, 2 and 3 respectively (Table S3, ESI†). In accordance with experimental observations, the barriers to oxidative addition relative to the mono-benzene adducts M[II]·C6H6 and M2[II]2·C6H6 are higher for rubidium in both monomeric (Rb, 24.9 kcal mol−1; Cs, 19.1 kcal mol−1) and dimeric (Rb, 28.3 kcal mol−1; Cs, 23.7 kcal mol−1) pathways. Preceding each optimised C–H activation transition state, is a Meisenheimer intermediate, A-M[II] or A-M2[II]2, the identification of which is consistent with existing computational studies of aromatic C–H activations by aluminyls. The dimeric products B-M2[II]2 (Rb, −3.7 kcal mol−1; Cs, −5.7 kcal mol−1) are exergonic and more stable than the corresponding monomers B-[II] (Rb, 8.9 kcal mol−1; Cs, 3.5 kcal mol−1), hence based on these DFT free energy values the oxidative product 3 is likely to resemble B-Cs2[II]. While the calculated local barriers in the Rb case do not preclude it from exhibiting benzene activation we have not observed any such activation under the experimental conditions used in the Cs case.
This work demonstrates the accessibility of [M{Al(NONDipp)}]2 aluminyls for each of the stable group 1 metals (Li–Cs). No reactivity between [Rb{Al(NONDipp)}]2 and benzene was observed, but oxidative cleavage of a C–H bond is enabled with [Cs{Al(NONDipp)}]2 hinting at a synergistic alkali metal effect.29
TXG and REM thank the EPSRC for financial support (EP/S029788/1) and Craig Irving for his wise advice regarding the NMR investigations. MPC and MJE acknowledge Government funding from the Marsden Fund Council, managed by Royal Society Te Apārangi (Grant Number: MFP-VUW2020). SEN and CLM acknowledge funding from the EPSRC (EP/R020752). MJE acknowledges a Victoria University of Wellington Doctoral Scholarship. This research used the Balena (Bath) and Rāpoi (VUW) High Performance Computing (HPC) Services. Data used within this publication can be accessed at https://doi.org/10.15129/e59234ad-9d5b-4fcd-8254-434f0870db11.
Conflicts of interest
There are no conflicts to declare.References
- T. X. Gentner and R. E. Mulvey, Angew. Chem., Int. Ed., 2021, 60, 9247–9262 CrossRef CAS PubMed.
- S. D. Robertson, M. Uzelac and R. E. Mulvey, Chem. Rev., 2019, 119, 8332–8405 CrossRef CAS PubMed.
- J. M. Gil-Negrete and E. Hevia, Chem. Sci., 2021, 12, 1982–1992 RSC.
- J. Hicks, P. Vasko, J. M. Goicoechea and S. Aldridge, Angew. Chem., Int. Ed., 2021, 60, 1702–1713 CrossRef CAS PubMed.
- R. J. Schwamm, M. D. Anker, M. Lein and M. P. Coles, Angew. Chem., Int. Ed., 2019, 58, 1489–1493 CrossRef CAS PubMed.
- R. J. Schwamm, M. P. Coles, M. S. Hill, M. F. Mahon, C. L. McMullin, N. A. Rajabi and A. S. S. Wilson, Angew. Chem., Int. Ed., 2020, 59, 3928–3932 CrossRef CAS PubMed.
- J. Hicks, P. Vasko, J. M. Goicoechea and S. Aldridge, Nature, 2018, 557, 92–95 CrossRef CAS PubMed.
- S. Kurumada, S. Takamori and M. Yamashita, Nat. Chem., 2020, 12, 36–39 CrossRef CAS PubMed.
- K. Koshino and R. Kinjo, J. Am. Chem. Soc., 2020, 142, 9057–9062 CrossRef CAS PubMed.
- R. J. Schwamm, M. S. Hill, H.-Y. Liu, M. F. Mahon, C. L. McMullin and N. J. Rajabi, Chem. – Eur. J., 2021, 27, 14971–14980 CrossRef CAS PubMed.
- S. Grams, J. Eyselein, J. Langer, C. Färber and S. Harder, Angew. Chem., Int. Ed., 2020, 59, 15982–15986 CrossRef CAS PubMed.
- M. J. Evans, M. D. Anker, M. G. Gardiner, C. L. McMullin and M. P. Coles, Inorg. Chem., 2021 DOI:10.1021/acs.inorgchem.1021c03012.
- J. Hicks, P. Vasko, J. M. Goicoechea and S. Aldridge, J. Am. Chem. Soc., 2019, 141, 11000–11003 CrossRef CAS PubMed.
- M. J. Evans, M. D. Anker, C. L. McMullin, S. E. Neale and M. P. Coles, Angew. Chem., Int. Ed., 2021, 60, 22289–22292 CrossRef CAS PubMed.
- M. Roy, J. Hicks, P. Vasko, A. Heilmann, A. M. Baston, J. Goicoechea and S. Aldridge, Angew. Chem., Int. Ed., 2021, 60, 22301–22306 CrossRef CAS PubMed.
- An alternative, low yielding route to the lithium aluminyl (XanthNONDipp)Al–Li(Et2O)2 has been reported. See ref. 15.
- T. X. Gentner, A. R. Kennedy, E. Hevia and R. E. Mulvey, ChemCatChem, 2021, 13, 2371–2378 CrossRef CAS.
- M. D. Anker, M. Lein and M. P. Coles, Chem. Sci., 2019, 10, 1212–1218 RSC.
- B. Cordero, V. Gómez, A. E. Platero-Prats, M. Revés, J. Echeverría, E. Cremades, F. Barragán and S. Alvarez, Dalton Trans., 2008, 2832–2838 RSC.
- S. F. McWilliams, K. R. Rodgers, G. Lukat-Rodgers, B. Q. Mercado, K. Grubel and P. L. Holland, Inorg. Chem., 2016, 55, 2960–2968 CrossRef CAS PubMed.
- M. Niemeyer and P. P. Power, Inorg. Chem., 1996, 35, 7264–7272 CrossRef CAS PubMed.
- M. Niemeyer and P. P. Power, Inorg. Chim. Acta, 1997, 263, 201–207 CrossRef CAS.
- P. S. V. Kumar, V. Raghavendra and V. Subramanian, J. Chem. Sci., 2016, 128, 1527–1536 CrossRef CAS.
- This classification is based on the criteria where the absolute potential density is less than twice the kinetic potential density (|V(r)| < 2G(r)), and when the Laplacian is greater than zero (∇2ρ(r) > 0). P. S. V. Kumar, V. Raghavendra and V. Subramanian, J. Chem. Sci., 2016, 2128, 1527–1536 CrossRef.
- M. Ziółkowski, S. J. Grabowski and J. Leszczynski, J. Phys. Chem. A, 2006, 110, 6514–6521 CrossRef PubMed.
- M. J. Evans, M. D. Anker and M. P. Coles, Inorg. Chem., 2021, 60, 4772–4778 CrossRef CAS PubMed.
- J. Hicks, P. Vasko, A. Heilmann, J. M. Goicoechea and S. Aldridge, Angew. Chem., Int. Ed., 2020, 59, 20376–20380 CrossRef CAS PubMed.
- S. Kurumada, K. Sugita, R. Nakano and M. Yamashita, Angew. Chem., Int. Ed., 2020, 59, 20381–20384 CrossRef CAS PubMed.
- N. Villegas-Escobar, A. Toro-Labbé and H. F. Schaefer III, Chem. – Eur. J., 2021, 27, 17369–17378 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Full experimental details and characterizing data; NMR spectra; details of crystallographic and computational experiments. CCDC 2111090 and 2111091. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1cc05379e |
| This journal is © The Royal Society of Chemistry 2022 |