
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Remote ortho-C–H functionalization via medium-sized cyclopalladation
Mario Martínez
Mingo
a,
Nuria
Rodríguez
 *ab,
Ramón Gómez
Arrayás
*ab and
Juan C.
Carretero
*ab
*ab,
Ramón Gómez
Arrayás
*ab and
Juan C.
Carretero
*ab
aDepartment of Organic Chemistry, Universidad Autónoma de Madrid (UAM), Facultad de Ciencias, Cantoblanco, 28049, Madrid, Spain. E-mail: n.rodriguez@uam.es; ramon.gomez@uam.es; juancarlos.carretero@uam.es
bInstitute for Advanced Research in Chemical Sciences (IAdChem), UAM, Madrid, Spain
First published on 27th January 2022
Abstract
Compared to the tremendous progress made in directed ortho-C–H functionalization via five- or six-membered cyclopalladation, protocols with the ability to selectively activate more remote C–H bonds through the intermediacy of larger, less favorable, seven- or eight-membered metalacycles are particularly challenging and remain rare. However, such a strategy would provide new retrosynthetic opportunities for generating structural diversity and complexity. Intense recent research based on the use of either mono-anionic bidentate or monodentate directing groups is characterizing this approach as an increasingly viable tool for selective C–C and C–X bond-forming reactions. This short review provides an overview of these strategies with an emphasis on mechanistic details, synthetic applicability, limitations, and key challenges.
Introduction
The direct catalytic transformation of C–H bonds into new functionalities offers a powerful and versatile means of generating structural diversity and complexity. The great potential, but also the challenges, associated to this strategy are due to the ubiquity of C–H bonds in organic substances.1–10 Despite the extensive progress made, achieving high site selectivity control is still a prominent goal in the field.11–16A successful approach towards this goal is the use of directing groups (DGs) to assist the C–H metalation step. A DG is a Lewis basic entity that acts as a ligand for the metal and brings the active catalytic species into close proximity to the desired C–H bond. This chelation control has a dual role in influencing local reactivity: (i) controlling the energy barrier for C–H bond activation and (ii) ensuring the desired selectivity by overriding inherent substrate preferences governed by electronic and/or steric bias. Furthermore, DGs with the ability to be easily removed or transformed allowing product diversification are of great value and actively sought after in this field.
In recent decades, directed C–H functionalization through metallacycle formation has been successfully employed in drug discovery and material science as well as pharmaceutical and chemical industries, among others.17–19 Nonetheless, the potential of this approach is mostly restricted to reactions that involve activation of proximal C–H bonds (generally beta or gamma to the directing group). In most of these cases, the kinetically preferred formation of five-membered metallacycles dictates selectivity, which is particularly true in the case of palladium catalysis.20 Five-membered metalation with PdII leads to the formation of square-planar complexes, whereas six or higher-membered palladacycles display less stable non-planar coordination.21
By contrast, remote functionalization of C–H bonds located distal to the DG remains undeveloped. A primary reason for this scarcity is that it requires the formation of metalacycles larger than 5- or 6-membered rings, which are kinetically and thermodynamically disfavored. In this context, the various DG-assisted methodologies that have appeared mainly involve the functionalization of C(sp2)–H bonds through six-membered palladacycles. Similarly, the meta- or para-selective functionalization of arene derivatives through a template-assisted palladation approach involving the formation of macropalladacycles with reduced ring strain (up to larger than 12 membered-rings) has also received significant attention.22–27 However, there is only a handful of protocols in the literature capable of achieving directed remote ortho-C–H functionalization via medium sized (7-9-membered) palladacycle intermediates, which highlights the challenging nature of this task. The majority of these examples involve seven-membered rings, with only two contributions proceeding via the intermediacy of eight-membered palladacycles (Scheme 1).
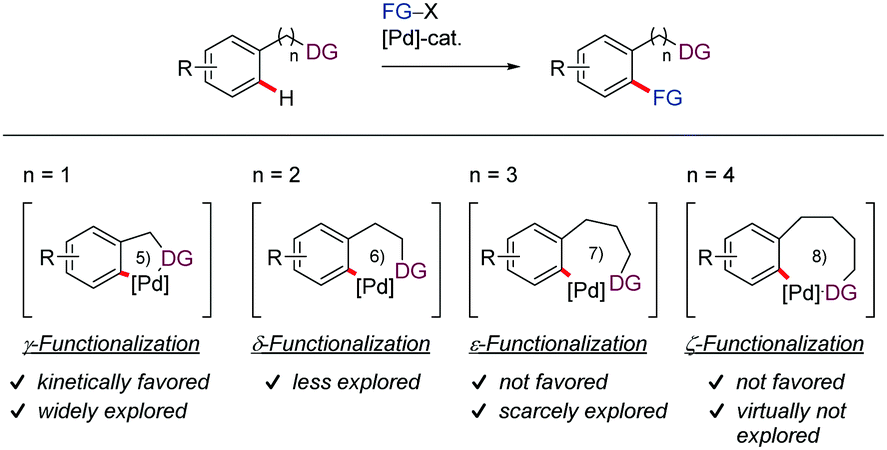 | ||
| Scheme 1 Five membered C(sp2)–H versus six, seven and eight membered C(sp2)–H metalation. | ||
The aim of this review is to bring together these remote ortho-directed C(sp2)–H bond transformations, through seven or eight-membered palladacycle intermediates, primarily emphasizing mechanistic aspects regarding site selectivity control. The aim of this overview is to stimulate impactful research in the field leading to practical solutions to the existing limitations and challenges. New strategies for selective functionalization of remote C–H bonds would provide new retrosynthetic opportunities for the construction of C–C and C–X bonds.
1. Formation of C–C bonds
Strategies involving C–C bond formation at inert C–H bonds have allowed the ready access to molecules that would otherwise be difficult to prepare, involving biologically relevant compounds, modern synthetic materials, and commodity chemicals.Among the variety of strategies described in the literature, the oxidative alkenylation of arenes (also known as Fujiwara–Moritani reaction or direct dehydrogenative Heck-coupling) is among the most prominent. This cross-coupling provides one of the most straightforward ways for the construction of styrene derivatives, which are very important building blocks in the chemical industry.28–33 Most of these catalytic C(sp2)–H transformations involve chelation-control through five- or six-membered palladacycles. The first example that was proposed to proceed through a seven-membered palladacycle was reported by Shi and Zhao in 2014. In this work, a highly efficient protocol for the Pd-catalyzed oxalylamide (OA)-directed mono-selective alkenylation of phenylpropylamine derivatives at ε position was devised (Scheme 2).34 Pd(OAc)2 (10 mol%) and Ag2CO3 in super-stoichiometric amount was found to be the best catalyst and oxidant combination, along with (n-BuO)2PO2H as additive in DCE at 120 °C for 24 h. This method was successfully applied to a wide range of olefins, such as terminal alkyl olefins, allyl alcohol derivatives, acrylaldehyde, allyl acetate, acrylates, and acrylonitrile (1–7). The scope of the reaction includes phenylpropylamine derivatives bearing different substitution patterns in the aromatic ring (8–16). In all cases studied, the methylene γ-C–H bonds, whose activation would involve five-membered cyclopalladation, remained unaltered. Furthermore, sequential double alkenylation at the two ortho positions with two different olefins provided a practical one-pot method for the synthesis of complex alkenylated arenes in good yields.
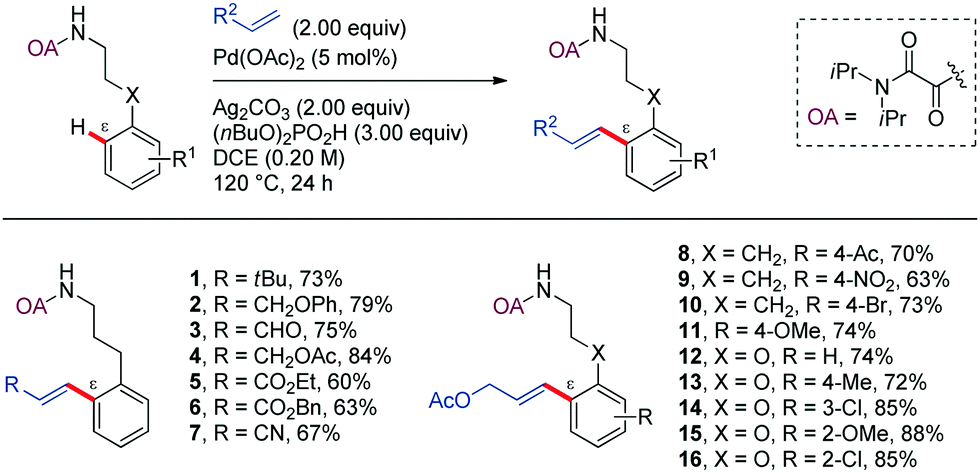 | ||
| Scheme 2 OA-Directed Pd-catalyzed alkenylation of arylpropyl amines with olefins. | ||
Mechanistic experiments in the presence of TEMPO, showing no detrimental effect in reactivity, argued against a radical pathway. The authors postulated a PdII/Pd0 mechanism where the (n-Bu)2PO2H may function as a basic ligand that facilitates the C–H activation step via a seven-membered ring palladacycle intermediate (Scheme 3). The resulting cyclopalladated complex would subsequently evolve through alkene insertion and β-hydride elimination to generate the alkenylated phenylpropylamine derivative along with Pd0. Reoxidation of the latter species by the silver salt would close the catalytic cycle. The authors suggested that the typical dialkenylation competing reaction was not favoured in this case due to a weak interaction between the palladium complex and the olefin.
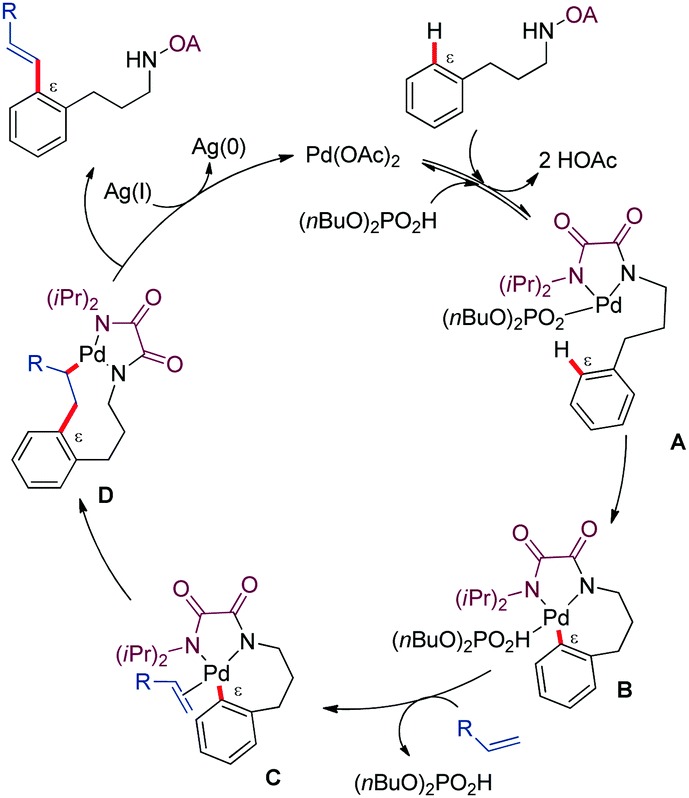 | ||
| Scheme 3 Mechanistic proposal for the alkenylation of phenylpropyl amine derivatives. | ||
Carretero reported in 2016 a suitable protocol for the Pd-catalyzed 2-pyridylsulfonyl (SO2Py) directed alkenylation of benzyl and phenethylsulfone derivatives with activated olefins (Scheme 4).35 In the case of benzyl sulfone derivatives (17–26), the metallacycle resulted from the C–H activation cleavage would be a seven-membered intermediate, coordinated by the N-atom from the pyridine moiety. On the other hand, phenethyl sulfone derivatives would lead to the challenging eight membered metalated intermediate (27–36), which has been scarcely investigated. In terms of reactivity, tether length of the DG did not significantly affect the yield of the product. Tolerance to a broad range of functional groups at both coupling partners, as well as to different substitution patterns on the aromatic ring are attractive features of this alkenylation protocol. In the case of phenethyl sulfone derivatives, substitution at the benzylic position is required to achieve high yields (27–36). Nevertheless, the stereochemical integrity of the stereogenic center at the benzylic position was preserved when using enantiomerically enriched substrates (27). This method was applied to the synthesis of indane derivatives holding three contiguous stereogenic centers upon by an intramolecular Michael-type addition of the α-sulfonyl carbanion of phenethyl sulfone derivatives to the newly introduced electrophilic alkene.
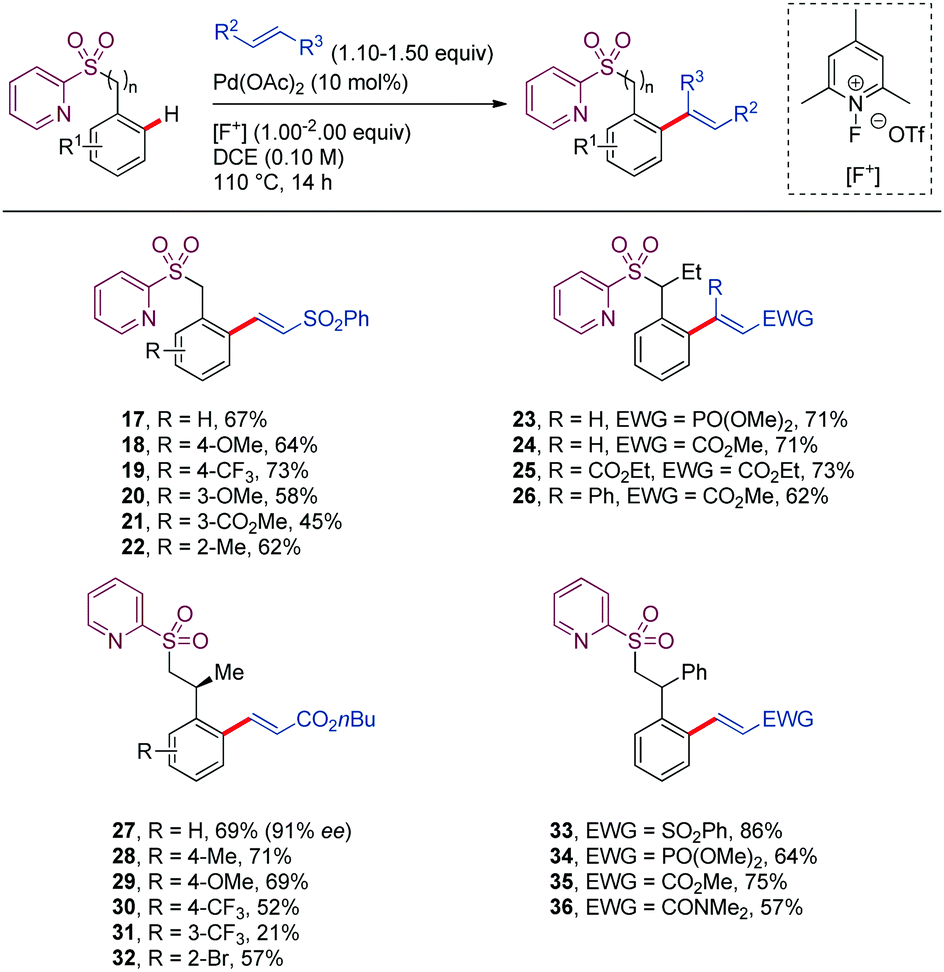 | ||
| Scheme 4 Pd-Catalyzed olefination of benzyl and phenethyl sulfone derivatives. | ||
Oxidative coupling of C–H bonds with alkynyl halides has become a convenient method for introducing an alkyne functionality into aromatic compounds.36–42 In most of the reported examples, a five-membered metallacycle is involved in the C–H activation process. However, in 2015, Zeng and Zhao reported the ability of the oxalyl amide (OA) as DG in promoting the ortho-alkynylation of phenethylamine and phenylpropylamine derivatives at δ and ε positions, respectively (Scheme 5).43 While the former functionalization presumably occurred through a six-membered palladacycle, the later implied the formation of a seven-membered ring analogue. Stoichiometric amounts of both AgOAc and KOAc were required to achieve high yields. The authors hypothesized that the acetate ion might act as a proton shuttle during the catalytic cycle, while the role of the silver ion, which was found to be crucial for ε- but not δ-derivatization remained unclear.
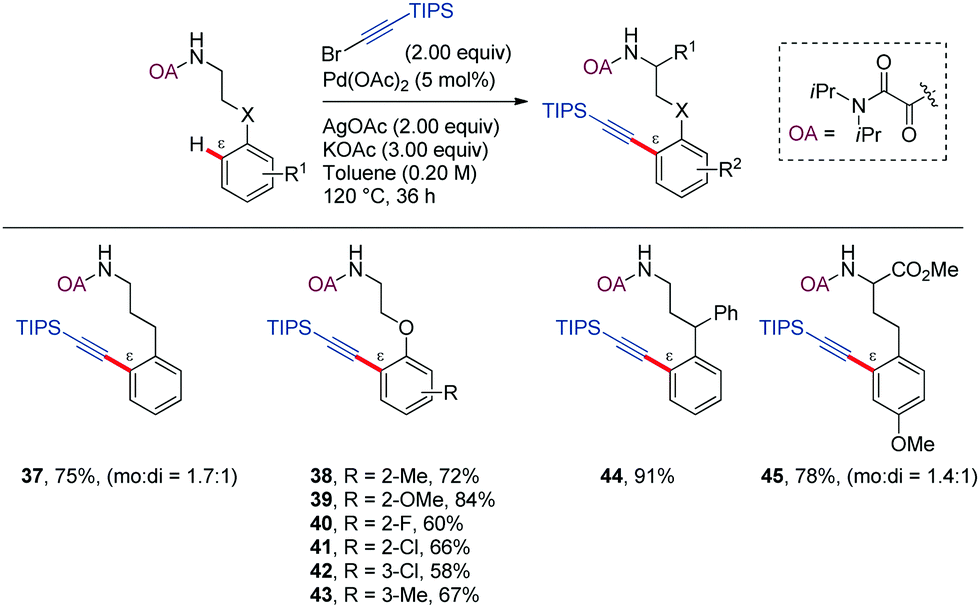 | ||
| Scheme 5 OA-Directed Pd-catalyzed alkynylation of arylpropyl amines with alkynyl halides. | ||
Regarding the scope, unsubstituted or para-substituted aromatic rings led to mixtures of mono and di-alkynylated products (37,45), while in the case of meta-substitution the reaction is preferred at the more sterically accessible C–H bond (42,43). Substitution at the benzylic position suppressed the formation of the di-alkynylated product, likely due to steric effects (44).
In 2019, we reported a practical method for the construction of benzazepinones through a Pd-catalyzed carbonylative cyclization of γ-arylpropylamine derivatives, including chiral α-amino acids. The use of the 2-pyridylsulfonamide (N-SO2Py) as DG was key for selectively targeting C(sp2)–H carbonylation at the remote ε-position. This method uses Mo(CO)6 as convenient substitute for CO gas. The functionalization of further positions failed, as well as the employ of different amide or sulfone based DGs, even the native amine (Scheme 6).44 A wide variety of substituted γ-arylpropylamine derivatives were used, noting that substrates bearing electron-rich aryl substituents provided higher reactivity (46–54). The applicability of this method to the derivatization of heteroarenes was showcased by the synthesis of a chiral analogue of rucaparib, a recently approved drug for ovarian cancer (55). Applications to late-stage modification of di- and tripeptides were also demonstrated (56,57).
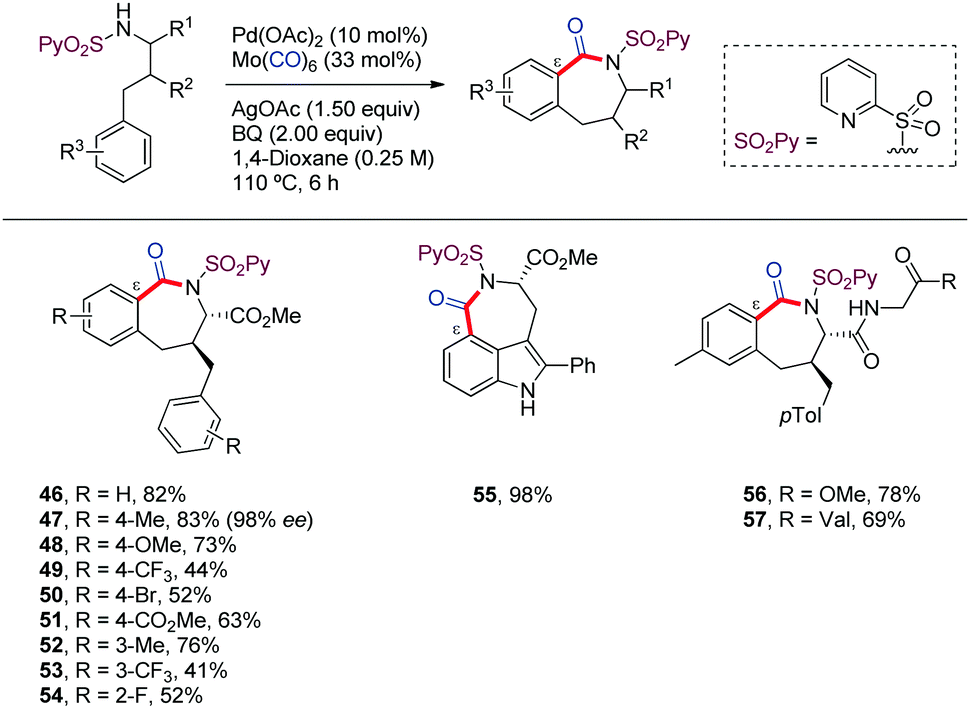 | ||
| Scheme 6 N-SO2Py-directed carbonylative cyclization of γ-arylpropyl amines. | ||
We proposed a PdII/Pd0 catalytic cycle initiated by coordination of the substrate to Pd(OAc)2 generating complex A′ in equilibrium with complex A (which serves as reversible off-cycle reservoir) (Scheme 7). Then, the PdII-catalyzed C–H activation would lead to complex B, which upon coordination to CO (C) followed by carbonyl insertion across the Pd–C bond could afford complex D. Reductive elimination would next release the benzazepinone product, along with reduced Pd0 species that would then be reoxidized back to the active PdII species via the combined action of BQ and AgOAc.
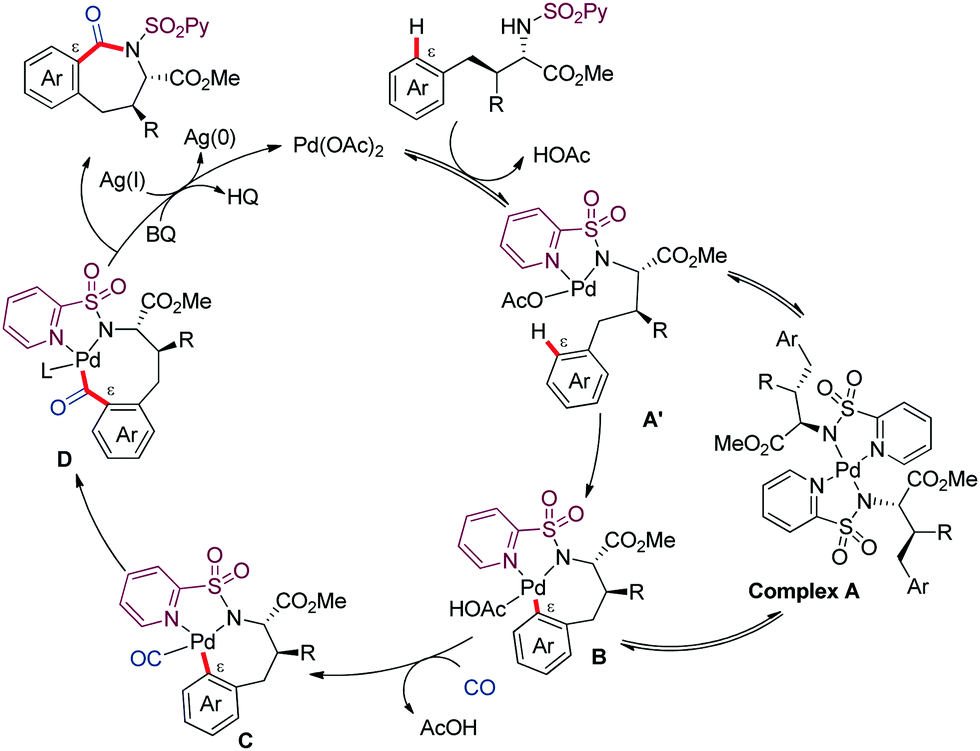 | ||
| Scheme 7 Mechanistic proposal for the ε-carbonylative cyclization. | ||
In the case of γ-aryl-substituted valine derivatives, which have available both aliphatic γ-C(sp3)–H (via a 5-membered palladacycle) and aromatic ε-C(sp2)–H bonds (via a 7-membered palladacycle), chemoselective reaction at C(sp2)–H bonds consistently predominated over activation of C(sp3)–H bonds (58–64) (Scheme 8). However, site-selectivity was found to be sensitive to the electronic properties of the aromatic ring, with aryl activation being more favored for substrates bearing electron-rich substituents. Theoretical and experimental mechanistic studies carried out subsequently by our research group revealed that the ε-C(sp2)–H bond cleavage in the γ-arylated valine substrate was significantly faster and more reversible than the γ-C(sp3)–H bond activation. However, the subsequent CO coordination from the C(sp3)–palladacycle led to more stable intermediates from which the reaction was irreversible.45 This knowledge provided the basis for the rational identification of reaction conditions that enabled reversing the selectivity from arene C(sp2)–H activation to C(sp3)–H cleavage, by promoting thermodynamic over kinetic control, thus leading to γ-lactam derivatives (65–71).45 This achievement is remarkable given the high selectivity for arene C–H activation over aliphatic C–H activation typically displayed by Pd.46–55
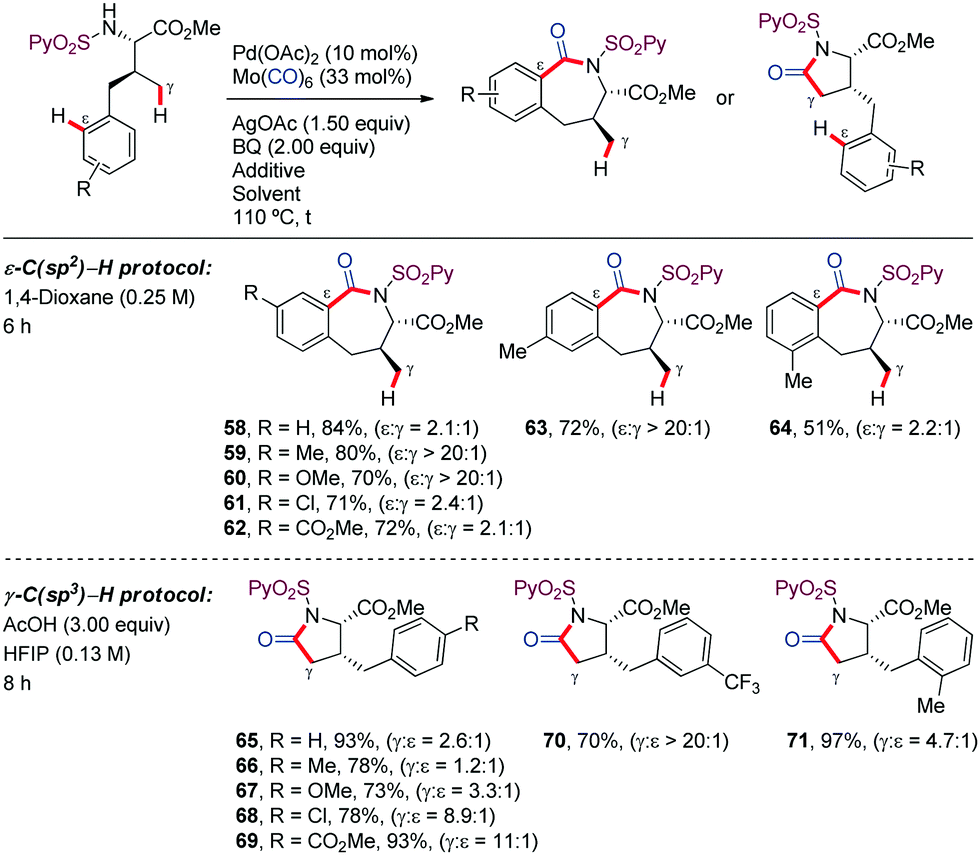 | ||
| Scheme 8 N-SO2Py-directed seven versus five membered functionalization. | ||
2. Formation of C–X bonds
2.1. C–N bond formation reactions
In a seminal contribution, Daugulis56 reported in 2012 the picolinamide (PA)-directed Pd-catalyzed oxidative cyclization of N-protected amines using PhI(OAc)2 as oxidant in toluene at 80–120 °C (Scheme 9a). This methodology allowed access to pyrrolidine, indoline, and isoindoline derivatives in good yields (72–75). The regioselectivity of the C–H activation was generally determined by the formation of a [5,6]-palladacycle intermediate. Subsequent oxidation to form high-valent palladium species, followed by C–N reductive elimination would lead to the reaction products. Interestingly, the formation of a dihydrophenanthridine from 2,6-diphenylbenzylamine, involving the construction of a six-membered ring, showed that cyclopalladation via a seven-membered ring is also feasible through this method (Scheme 9b). However, only a single example of this type of ε-functionalization was disclosed (76).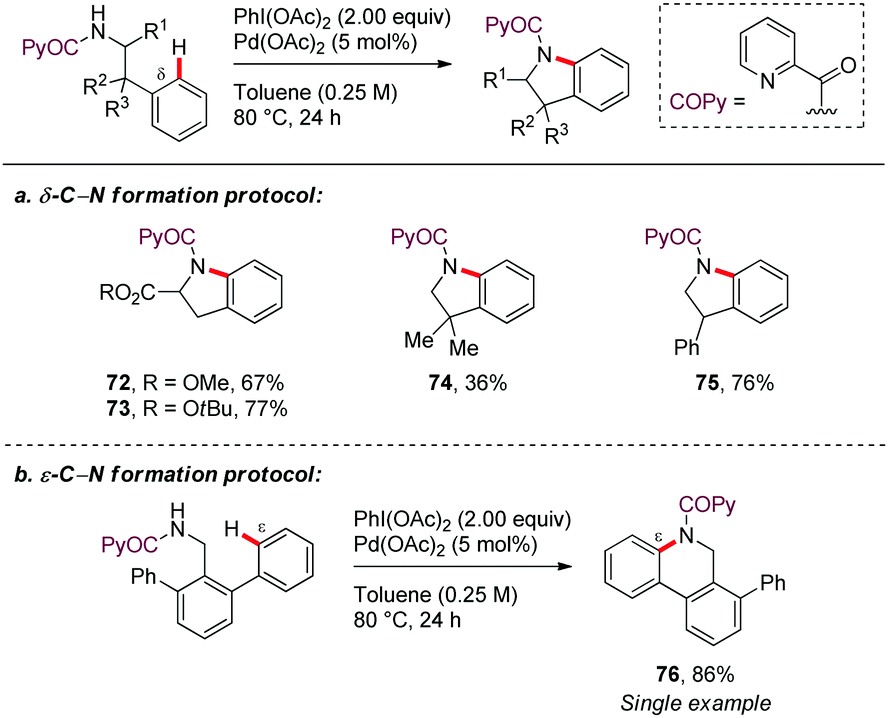 | ||
| Scheme 9 PA-Directed formation of dihydrophenanthridine derivatives. | ||
Shortly after, Chen demonstrated the generality of this direct C–H/N–H oxidative cyclization by developing a two-step method for the synthesis of phenanthridines from easily accessible PA-protected benzylamine derivatives (Scheme 10).57 In the first step, an improved protocol for the Pd-catalyzed PA-directed ortho-C–H arylation with iodoarenes was devised that avoids using expensive silver additives. Next, the intramolecular dehydrogenative coupling was achieved under Pd-catalysis using PhI(OAc)2 and Cu(OAc)2 as oxidants, affording the corresponding phenanthridine derivatives (77–83). The ortho-selectivity in the first step is dictated by a five-membered palladacycle intermediate, whereas the intermediacy of a seven-membered metallacycle is assumed for controlling ε-selectivity in the second step. In general, electron-rich arene motifs provided better yields than electron-deficient substrates.
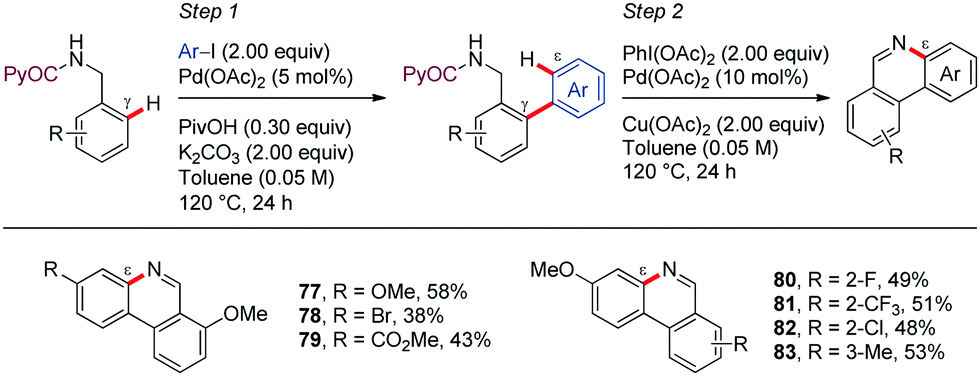 | ||
| Scheme 10 PA-Directed formation of phenanthridine derivatives. | ||
In 2014, Yao and Zhao58 reported a novel protocol for the Pd-catalyzed ε-selective intramolecular C(sp2)–H amination of γ-arylpropylamine derivatives using an oxalyl amide as N,N-bidentate DG (Scheme 11). Tetrahydroquinoline and benzomorpholine derivatives were efficiently assembled in good yields in the presence of PhI(OAc)2 as oxidant under low catalyst loading (3 mol%) and mild conditions (60 °C), thus enabling wide group tolerance (84–98).
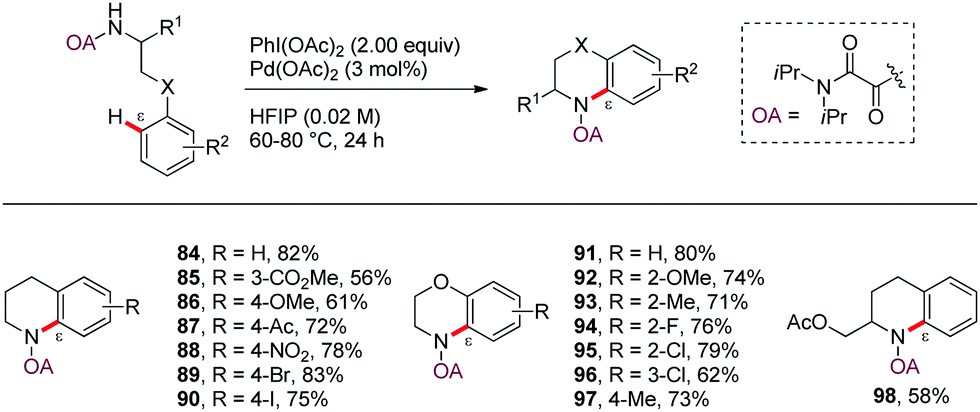 | ||
| Scheme 11 Oxalyl amide directed Pd-catalyzed intramolecular amination of arylpropyl amines. | ||
H/D scrambling studies performed in the absence of PhI(OAc)2 revealed a high H/D exchange at the ε-position (99-D, 62% H/D scrambling) but not at the γ-methylene position, suggesting that the PdII-catalyzed aryl C–H bond cleavage is reversible (Scheme 12).
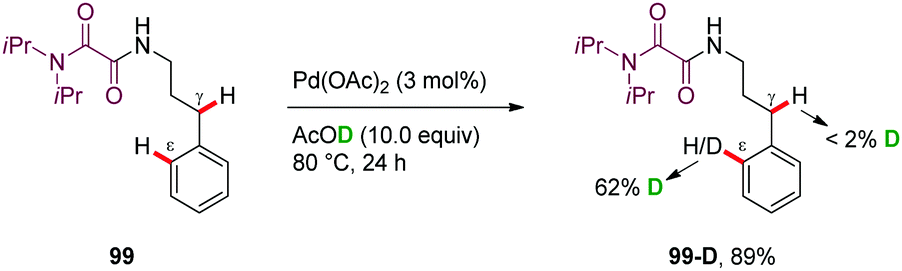 | ||
| Scheme 12 H/D scrambling experiment on OA-directed ε-C–H activation. | ||
2.2. C–O bond formation reactions
C–O bond formation, especially acetoxylation, has received substantial attention driven by the importance of the acetoxy group in organic synthesis. An additional complication inherent to any remote C–H oxygenation process of amine derivatives is how to prevent the competitive intramolecular C–H amination leading to the kinetically and thermodynamically favoured cyclized products. In many cases, the formation of the intermolecular C–H oxygenation or intramolecular C–H amination product depends on the substrate.59–62The first example of a remote C–H oxygenation reaction assumed to presumably proceed through a seven-membered palladacycle was reported by Yu in 2010 (Scheme 13).63 In this work, the ortho-C–H acetoxylation of phenlyethyltriflamides, likely via six-membered ring cyclopalladation, is described using tert-butyl peroxyacetate as the stoichiometric oxidant and either DMF or CH3CN as the promoter. However, an isolated example on the ε-acetoxylation of a γ-phenylpropylamine derivative, having a one atom longer tether, was reported, showcasing the feasibility of extending this method to the intermediacy of seven-membered palladacycles. Nevertheless, the yield of the isolated product remained low (100).
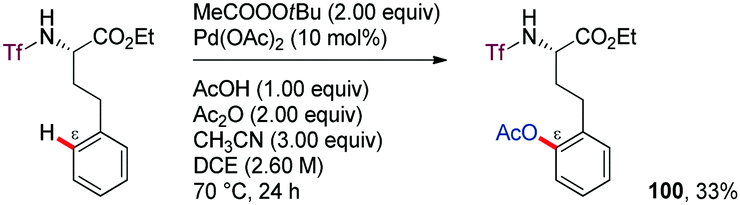 | ||
| Scheme 13 ortho-Acetoxylation of a triflamide derivative. | ||
Along similar lines, Babu64 reported in 2016 a chemoselective Pd-catalyzed ortho-acetoxylation of PA-protected (furan-2-yl)- or (thien-2-yl)methanamine derivatives with PhI(OAc)2 (101–110) (Scheme 14). Among the bidentate DGs tested, picolinamide (PA) and pyrazine-2-carboxamide (PyrA) were the most effective in the ε-C–H acetoxylation (111), whereas the oxalylamide (OA) showed lower efficiency (112). However, removal of the bidentate DG from the acetoxylated biaryl systems was found to be difficult. Interestingly, when a 2-(thien-2-yl)benzylamine derivative having the complementary biaryl system was subjected to identical reaction conditions, the oxidative cyclization product was obtained instead of the ε-C–H acetoxylated product (113) (Scheme 15). This result highlights that this transformation is strongly substrate dependent.
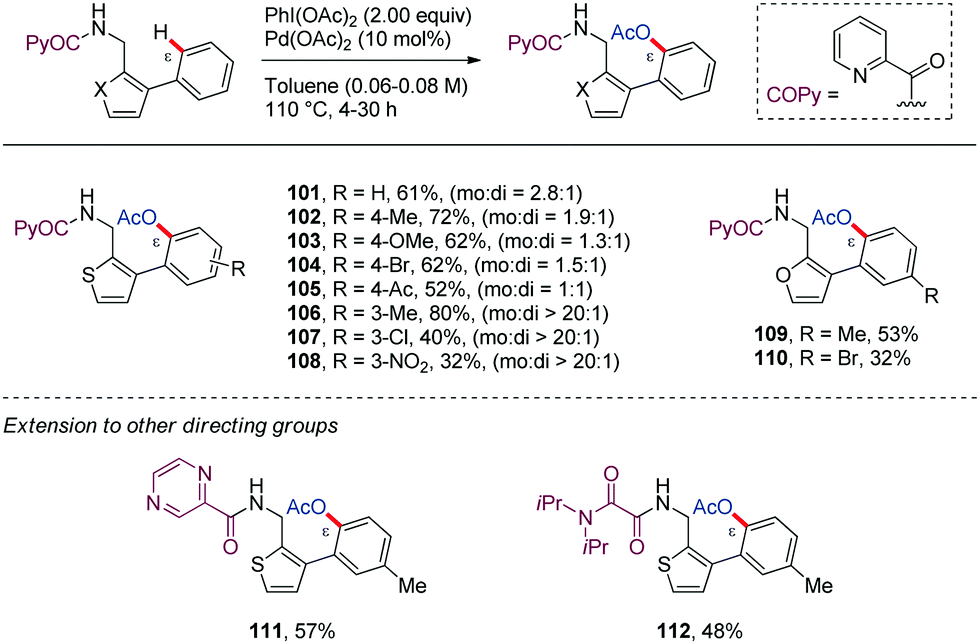 | ||
| Scheme 14 Pd-Catalyzed ε-acetoxylation of heteroaryl–aryl type biaryl systems. | ||
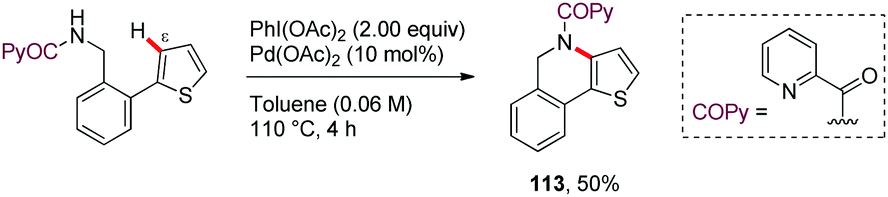 | ||
| Scheme 15 ε-C–N Bond formation of 2-thiophenyl-benzyl picolinamide derivative. | ||
The authors tentatively suggested a PdII/PdIV catalytic pathway (Scheme 16). Bidentate coordination of the DG-linked substrate and subsequent C–H bond cleavage would lead to a metalated [5,7]-bycyclic intermediate (A). Subsequent oxidation to PdIV species type-B by PhI(OAc)2, followed by reductive elimination would afford the ε-acetoxylated product along with PdII.
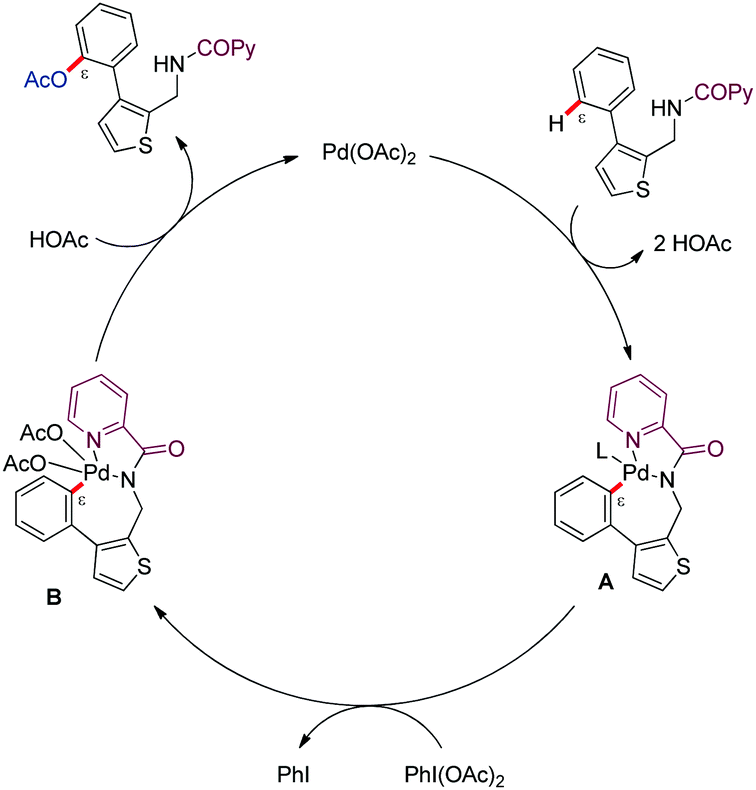 | ||
| Scheme 16 Mechanistic proposal for the acetoxylation of biaryl systems. | ||
2.3. C–I bond formation reactions
Iodination of aryl C–H bonds provides a straightforward access to synthetically valuable building blocks. In 2008, Yu65 described the iodination of aryl C–H bonds located four, five or even six bonds away from the oxazoline DG using Pd(OAc)2 as the catalyst and IOAc as both oxidant and iodine source (114–118) (Scheme 17).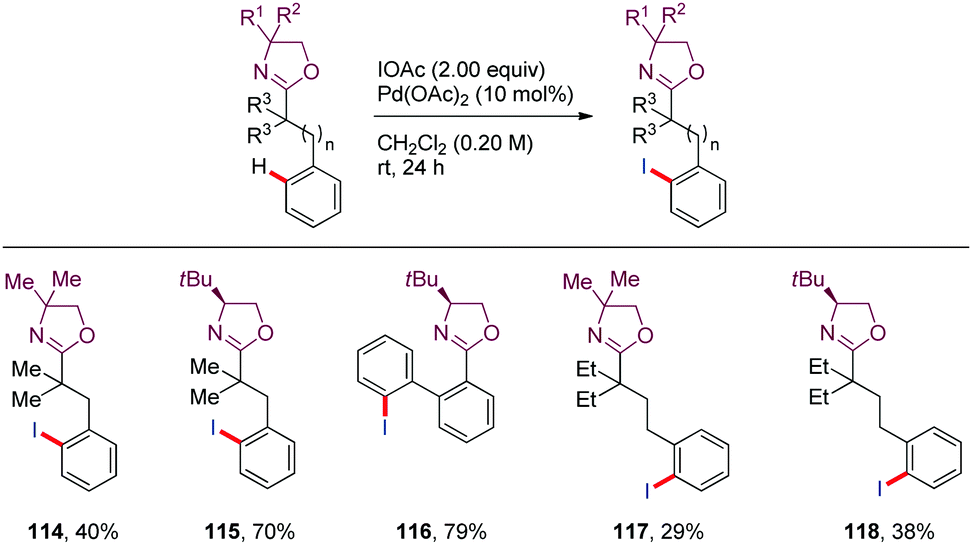 | ||
| Scheme 17 Iodination of oxazoline derivatives. | ||
Systematic kinetic isotope studies revealed a mechanistic regime shift as the tether length of the DG to the target C–H bond increases. The authors tentatively proposed that the functionalization of nearby C–H bonds proceeded via an oxidative addition mechanism or a σ-bond metathesis mechanism (KIE = 3.5) whereas the more remote C–H bond activation seemed to occur through an electrophilic palladation pathway based on both isotope (KIE = 1.5) and electronic effects observed. In the latter cases, the initial palladation appears to be slower than the C–H bond cleavage.
Summary and outlook
In the past decades, a variety of DGs have been designed for controlling the chelate-assisted direct functionalization of arenes mainly through either five- or six-membered palladacycle intermediates. These new methods have found application in the synthesis of a wide range of functionalized molecules and bioactive compounds. However, the successful use of DGs capable of selectively facilitating the direct activation of more remotely located C–H bonds remains an outstanding challenge. Most of the few examples reported in the literature involve the remote ortho-C(sp2)–H bond functionalization of amine derivatives using mono-anionic bidentate DGs such as oxalylamide (OA), picolinamide (PA) and N-(2-pyridyl)sulfonamide (N-SO2Py), whereas two contributions were reported relying on the use of monodentate DGs: oxazoline and 2-pyridylsulfone derivatives. Therefore, there is still plenty room for further development in terms of both designing novel powerful and versatile DGs or discovering new types of reactivity leading to remote functionalization.Regarding the metal source, Pd-catalysis has dominated this field. Hence, as in all fields of metal catalysis, the quest for switching from palladium to other metals, especially first-row transition metals, is a relevant driving force for innovation.
At a more fundamental level, a remaining challenge in this area is to gain an understanding of the factors that govern the regioselectivity of competing C–H functionalization. This basic knowledge would undoubtedly contribute to design new protocols discriminating about accessible C–H bonds present in the organic framework.
We hope that this review can contribute to stimulating new perspectives and innovative strategies in the remote directed ortho-C(sp2)–H functionalization via catalytic cyclometalation.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the Ministerio de Ciencia e Innovación (MICINN) (Agencia Estatal de Investigación/Project PGC2018-098660-B-I00) and Fondo Europeo de Desarrollo Regional (FEDER, UE), for financial support. M. M.-M. thanks MINECO for a FPI predoctoral fellowship.Notes and references
- N. Kuhl, M. N. Hopkinson, J. Wencel-Delord and F. Glorius, Angew. Chem., Int. Ed., 2012, 51, 10236–10254 CrossRef CAS PubMed.
- S. De Sarkar, W. Liu, S. I. Kozhushkov and L. Ackermann, Adv. Synth. Catal., 2014, 356, 1461–1479 CrossRef CAS.
- F. Zhang and D. R. Spring, Chem. Soc. Rev., 2014, 43, 6906–6919 RSC.
- O. Daugulis, J. Roane and L. D. Tran, Acc. Chem. Res., 2015, 48, 1053–1064 CrossRef CAS PubMed.
- S. De Sarkar, Angew. Chem., Int. Ed., 2016, 55, 10558–10560 CrossRef CAS PubMed.
- J. C. K. Chu and T. Rovis, Angew. Chem., Int. Ed., 2018, 57, 62–101 CrossRef CAS PubMed.
- T. Sawano and H. Yamamoto, J. Org. Chem., 2018, 83, 4889–4904 CrossRef CAS PubMed.
- G. Meng, N. Y. S. Lam, E. L. Lucas, T. G. Saint-Denis, P. Verma, N. Chekshin and J.-Q. Yu, J. Am. Chem. Soc., 2020, 142, 10571–10591 CrossRef CAS PubMed.
- S. Rej, Y. Ano and N. Chatani, Chem. Rev., 2020, 120, 1788–1887 CrossRef CAS PubMed.
- N. Y. S. Lam, K. Wu and J.-Q. Yu, Angew. Chem., Int. Ed., 2021, 15767–15790 CrossRef CAS PubMed.
- J. Wencel-Delord and F. Glorius, Nat. Chem., 2013, 5, 369–375 CrossRef CAS PubMed.
- T. Gensch, M. N. Hopkinson, F. Glorius and J. Wencel-Delord, Chem. Soc. Rev., 2016, 45, 2900–2936 RSC.
- J. F. Hartwig and M. A. Larsen, ACS Cent. Sci., 2016, 2, 281–292 CrossRef CAS PubMed.
- D. J. Abrams, P. A. Provencher and E. J. Sorensen, Chem. Soc. Rev., 2018, 47, 8925–8967 RSC.
- M. J. Caplin and D. J. Foley, Chem. Sci., 2021, 12, 4646–4660 RSC.
- S. R. Neufeldt and M. S. Sanford, Acc. Chem. Res., 2012, 45, 936–946 CrossRef CAS PubMed.
- K. Chen and X. Lei, Curr. Opin. Green Sustainable Chem., 2018, 11, 9–14 CrossRef.
- D. J. Abrams, P. A. Provencher and E. J. Sorensen, Chem. Soc. Rev., 2018, 47, 8925–8967 RSC.
- O. Baudoin, Angew. Chem., Int. Ed., 2020, 59, 17798–17809 CrossRef CAS PubMed.
- V. K. Jain, Coord. Chem. Rev., 2021, 427, 213546 CrossRef CAS.
- Y.-F. Yang, G. Chen, X. Hong, J.-Q. Yu and K. N. Houk, J. Am. Chem. Soc., 2017, 139, 8514–8521 CrossRef CAS PubMed.
- J. Yang, Org. Biomol. Chem., 2015, 13, 1930–1941 RSC.
- A. Dey, S. Agasti and D. Maiti, Org. Biomol. Chem., 2016, 14, 5440–5453 RSC.
- J. Li, S. De Sarkar and L. Ackermann, in C–H Bond Activation and Catalytic Functionalization I, ed. P. H. Dixneuf and H. Doucet, Springer International Publishing, Cham, 2016, pp. 217–257 Search PubMed.
- M. T. Mihai, G. R. Genov and R. J. Phipps, Chem. Soc. Rev., 2018, 47, 149–171 RSC.
- A. Dey, S. K. Sinha, T. K. Achar and D. Maiti, Angew. Chem., Int. Ed., 2019, 58, 10820–10843 CrossRef CAS PubMed.
- S. Sasmal, U. Dutta, G. K. Lahiri and D. Maiti, Chem. Lett., 2020, 49, 1406–1420 CrossRef CAS.
- J. Le Bras and J. Muzart, Chem. Rev., 2011, 111, 1170–1214 CrossRef CAS PubMed.
- C. S. Yeung and V. M. Dong, Chem. Rev., 2011, 111, 1215–1292 CrossRef CAS PubMed.
- L. Zhou and W. Lu, Chem. – Eur. J., 2014, 20, 634–642 CrossRef CAS PubMed.
- W. Ma, P. Gandeepan, J. Li and L. Ackermann, Org. Chem. Front., 2017, 4, 1435–1467 RSC.
- P. Gandeepan and L. Ackermann, C–H Activation for Asymmetric Synthesis, John Wiley & Sons, Ltd, 2019, pp. 239–274 Search PubMed.
- W. Ali, G. Prakash and D. Maiti, Chem. Sci., 2021, 12, 2735–2759 RSC.
- Q. Wang, J. Han, C. Wang, J. Zhang, Z. Huang, D. Shi and Y. Zhao, Chem. Sci., 2014, 5, 4962–4967 RSC.
- P. D. Legarda, A. García-Rubia, R. Gómez-Arrayás and J. C. Carretero, Adv. Synth. Catal., 2016, 358, 1065–1072 CrossRef CAS.
- I. V. Seregin, V. Ryabova and V. Gevorgyan, J. Am. Chem. Soc., 2007, 129, 7742–7743 CrossRef CAS PubMed.
- Y. Ano, M. Tobisu and N. Chatani, J. Am. Chem. Soc., 2011, 133, 12984–12986 CrossRef CAS PubMed.
- Y.-J. Liu, Y.-H. Liu, S.-Y. Yan and B.-F. Shi, Chem. Commun., 2015, 51, 6388–6391 RSC.
- L. D. Caspers, P. Finkbeiner and B. J. Nachtsheim, Chem. – Eur. J., 2017, 23, 2748–2752 CrossRef CAS PubMed.
- G. Li, P. Liu, J. Zhang, D.-Q. Shi and Y. Zhao, Org. Chem. Front., 2017, 4, 1931–1934 RSC.
- B. Liu, W. Ouyang, J. Nie, Y. Gao, K. Feng, Y. Huo, Q. Chen and X. Li, Chem. Commun., 2020, 56, 11255–11258 RSC.
- B. S. Schreib, M. Fadel and E. M. Carreira, Angew. Chem., Int. Ed., 2020, 59, 7818–7822 CrossRef CAS PubMed.
- M. Guan, C. Chen, J. Zhang, R. Zeng and Y. Zhao, Chem. Commun., 2015, 51, 12103–12106 RSC.
- M. Martínez-Mingo, N. Rodríguez, R. GómezArrayás and J. C. Carretero, Org. Lett., 2019, 21, 4345–4349 CrossRef PubMed.
- M. Martínez-Mingo, I. Alonso, N. Rodríguez, R. G. Arrayás and J. C. Carretero, Catal. Sci. Technol., 2021, 11, 1590–1601 RSC.
- L.-C. Campeau, D. J. Schipper and K. Fagnou, J. Am. Chem. Soc., 2008, 130, 3266–3267 CrossRef CAS PubMed.
- D. J. Schipper, L.-C. Campeau and K. Fagnou, Tetrahedron, 2009, 65, 3155–3164 CrossRef CAS.
- P. Novák, A. Correa, J. Gallardo-Donaira and R. Martin, Angew. Chem., Int. Ed., 2011, 50, 12236–12239 CrossRef PubMed.
- F.-L. Zhang, K. Hong, T.-J. Li, H. Park and J.-Q. Yu, Science, 2016, 351, 252–256 CrossRef CAS PubMed.
- F. Ma, M. Lei and L. Hu, Org. Lett., 2016, 18, 2708–2711 CrossRef CAS PubMed.
- H. Park, K. Yoo, B. Jung and M. Kim, Tetrahedron, 2018, 74, 2048–2055 CrossRef CAS.
- Q. He, Y. Ano and N. Chatani, Chem. Commun., 2019, 55, 9983–9986 RSC.
- H. Park, P. Verma, K. Hong and J.-Q. Yu, Nat. Chem., 2018, 10, 755–762 CrossRef CAS PubMed.
- G. Hong, P. D. Nahide, U. K. Neelam, P. Amadeo, A. Vijeta, J. M. Curto, C. E. Hendrick, K. F. VanGelder and M. C. Kozlowski, ACS Catal., 2019, 9, 3716–3724 CrossRef CAS PubMed.
- T. Gogula, J. Zhang, M. R. Lonka, S. Zhang and H. Zou, Chem. Sci., 2020, 11, 11461–11467 RSC.
- E. T. Nadres and O. Daugulis, J. Am. Chem. Soc., 2012, 134, 7–10 CrossRef CAS PubMed.
- R. Pearson, S. Zhang, G. He, N. Edwards and G. Chen, Beilstein J. Org. Chem., 2013, 9, 891–899 CrossRef CAS PubMed.
- C. Wang, C. Chen, J. Zhang, J. Han, Q. Wang, K. Guo, P. Liu, M. Guan, Y. Yao and Y. Zhao, Angew. Chem., Int. Ed., 2014, 53, 9884–9888 CrossRef CAS PubMed.
- B. V. S. Reddy, L. R. Reddy and E. J. Corey, Org. Lett., 2006, 8, 3391–3394 CrossRef CAS PubMed.
- F.-R. Gou, X.-C. Wang, P.-F. Huo, H.-P. Bi, Z.-H. Guan and Y.-M. Liang, Org. Lett., 2009, 11, 5726–5729 CrossRef CAS PubMed.
- Q. Li, S.-Y. Zhang, G. He, W. A. Nack and G. Chen, Adv. Synth. Catal., 2014, 356, 1544–1548 CrossRef CAS.
- G. He, G. Lu, Z. Guo, P. Liu and G. Chen, Nat. Chem., 2016, 8, 1131–1136 CrossRef CAS.
- C. J. Vickers, T.-S. Mei and J.-Q. Yu, Org. Lett., 2010, 12, 2511–2513 CrossRef CAS PubMed.
- Naveen, V. Rajkumar, S. A. Babu and B. Gopalakrishnan, J. Org. Chem., 2016, 81, 12197–12211 CrossRef CAS PubMed.
- J.-J. Li, R. Giri and J.-Q. Yu, Tetrahedron, 2008, 64, 6979–6987 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2022 |