
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceExploring the dynamic nature of divalent metal ions involved in DNA cleavage by CRISPR–Cas12a†
Heyjin
Son‡
a,
Jaeil
Park‡
ab,
You Hee
Choi
ac,
Youngri
Jung
d,
Joong-Wook
Lee
b,
Sangsu
Bae
d and
Sanghwa
Lee
 *a
*a
aAdvanced Photonics Research Institute, Gwangju Institute of Science and Technology, Gwangju 61005, Republic of Korea. E-mail: sanglee@gist.ac.kr
bDepartment of Physics and Optoelectronics Convergence Research Center, Chonnam National University, Gwangju 61186, Republic of Korea
cMinistry of Food and Drug Safety (MFDS), Cheongju 28159, Republic of Korea
dDepartment of Chemistry, Hanyang University, Seoul 04763, Republic of Korea
First published on 14th January 2022
Abstract
CRISPR–Cas12a has been widely used in genome editing and nucleic acid detection. In both of these applications, Cas12a cleaves target DNA in a divalent metal ion-dependent manner. However, when and how metal ions contribute to the cleavage reaction is unclear. Here, using a single-molecule FRET assay, we reveal that these metal ions are necessary for stabilising cleavage-competent conformations and that they are easily exchangeable, suggesting that they are dynamically coordinated.
CRISPR–Cas12a, an RNA-guided double-stranded DNA (dsDNA) endonuclease, has received considerable attention owing to its powerful ability for genome editing and nucleic acid detection.1–5 To cleave target DNA, Cas12a first unwinds the dsDNA target known as R-loop formation with the crRNA complementary to the target DNA and sequentially catalyses the DNA strands by a single catalytic site in the RuvC domain.6–8 Recently, in the Cas12i2 R-loop complex, the coordination of two Mg2+ ions interacting with essential residues (DDE) in the catalytic pocket of the RuvC domain has been resolved by crystal structure.9 These core residues are highly conserved in a variety of type V CRISPR–Cas12 enzymes, and mutations in the residues or the absence of divalent metal ions abolishes the nuclease activity of Cas12 enzymes.10–12 Furthermore, a recent study reported that, in the presence of diverse divalent metal ions, Cas12a enzymes possess indiscriminate single-stranded DNase activity.13
Despite the important role of divalent metal ions in the DNA cleavage reaction, little is known about when and how the metal ions are specifically involved in individual reaction steps of Cas12a. In this study, we used single-molecule fluorescence resonance energy transfer (FRET) assay to explore how the metal ions facilitate DNA cleavage by Cas12a. Single-molecule FRET has been widely used to study biomolecular interactions with sub-nanometer sensitivity owing to its ability to monitor distance changes between a donor and an acceptor fluorophore in real time. Similar methodological approaches recently have been harnessed to observe the molecular interactions between Cas12a ribonucleoprotein (RNP) and target DNA during the DNA cleavage reaction.7,14–17 Here we found that the double stranded DNA cleavage reaction proceeds via divalent cation-dependent manner. Particularly, the CRISPR–Cas12a R-loop complex, a prerequisite for DNA cleavage, was highly stabilised as the concentration of the divalent cation increased, implying allosteric activation of the enzyme by the divalent metal ions. Using a real-time buffer exchange experiment, we directly examined the exchangeability of the divalent metal ions in the Cas12a R-loop complex.
To monitor the individual reaction steps of Cas12a using the single-molecule FRET assay, we prepared a Cy5 (acceptor)-labelled crRNA and double-stranded target DNA labelled with Cy3 (donor) and biotin. The target DNA was immobilised on the polymer-coated quartz surface of the detection chamber using biotin–streptavidin interaction. The preassembled Cas12a–crRNA complexes were subsequently introduced into the detection chamber with the designated concentration of divalent metal ions while recoding the fluorescence using total internal reflection fluorescence (TIRF) microscopy (Fig. 1A).
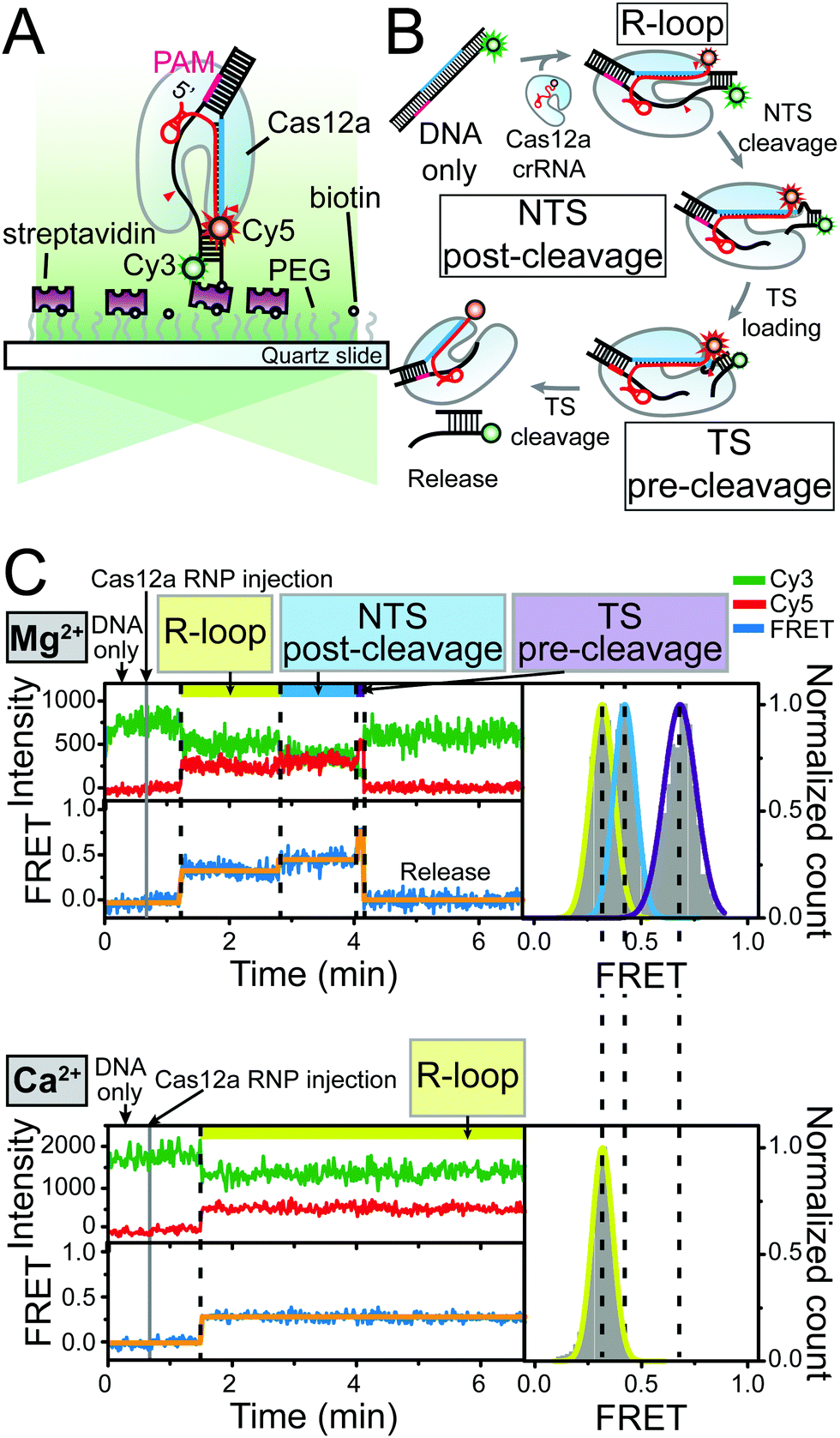 | ||
| Fig. 1 Mg2+-dependent DNA cleavage reaction by Cas12a RNP. (A) Schematic diagram of single-molecule FRET experiment to monitor the DNA cleavage reaction by Cas12a RNP in real-time. (B) Illustrative diagram of the process of DNA target cleavage by Cas12a RNP. Cy3 (donor) and Cy5 (acceptor) were depicted as green and red spheres, respectively. (C) Representative time traces of Cy3 (green) and Cy5 (red) fluorescence intensity and corresponding FRET efficiency (blue) in the presence of 20 nM Cas12a RNP and 10 mM Mg2+ (upper panel) or 10 mM Ca2+ (lower panel) ions. Orange lines were added to the FRET time trace as an eye guide to visualize the transition clearly. Corresponding FRET histograms of individual reaction steps were shown in the right panel of each time trace. Each FRET histogram was fitted to a single Gaussian function and the fitted lines were coloured in the same colour scheme used above the time trace to indicate each reaction step. | ||
We first investigated the conformational dynamics of the Cas12a complex under different reaction conditions (Mg2+ or Ca2+), respectively. In the presence of 10 mM Mg2+, we successfully obtained time traces of the fluorescence intensity and FRET efficiency and corresponding FRET histograms that represent the sequential double-stranded DNA cleavage reaction by the Cas12a RNP complex beginning with the non-target strand (NTS) and progressing to the target strand (TS), as previously reported (Fig. 1B and C).7 With a high concentration of Mg2+ ions (10 mM), the double-stranded DNA cleavage reaction proceeded via three distinct reaction steps after the binding of Cas12a RNP: the NTS pre-cleavage (R-loop), NTS post-cleavage, and TS pre-cleavage states. In the presence of 10 mM Ca2+ ions, however, the Cas12a R-loop complex was stably established but the target DNA was not cleaved. This loss of cleavage activity of Cas12a RNP under reaction conditions that include Ca2+ ions was also observed in the in vitro cleavage assay, which is in agreement with previous biochemical studies (Fig. S1, ESI†).18 In addition, even in the NTS pre-nicked DNA, a structurally stable but catalytically inactive state was observed in the Ca2+ buffer (Fig. S2, ESI†). These results indicate that, although Ca2+ ions stabilise the Cas12a RNP–DNA complexes, they probably inhibit DNA scission. In addition to these two divalent cations, we examined the effect of various divalent metal ions on the DNA cleavage reaction by Cas12a (Fig. S3, ESI†).
To examine the stable Cas12a R-loop complex formation process in connection with divalent metal ions in further detail, we performed Ca2+ titration experiments (Fig. 2A and Fig. S4, ESI†). Representative time traces of donor and acceptor intensities and corresponding dwell-time histograms for the R-loop state are shown in Fig. 2A. As the Ca2+ concentration increased, the R-loop complex in the NTS pre-cleavage state was dramatically stabilised. Even when the Ca2+ concentration was as low as 0.9 mM, Cas12a RNPs did not dissociate for up to 30 min and the R-loop complex was robustly maintained (Fig. 2B). Furthermore, in our recent paper, we obtained similar results even in the Mg2+ titration experiment.19 In the case of Mg2+ buffer condition, the stabilisation of the Cas12a R-loop complex by Mg2+ ions consequently promoted DNA cleavage (Fig. S6, ESI†). This finding indicates that divalent metal ions play a structurally important role in the Cas12a-mediated DNA cleavage reaction by stabilising the R-loop complex for cleavage-competent state.
 | ||
| Fig. 2 Stabilisation of the Cas12a R-loop complex by divalent metal ions. (A) Representative Cy3 (green) and Cy5 (red) fluorescence intensity time trace showing the formation of the Cas12a R-loop complex induced by divalent metal ions in the presence of 20 nM Cas12a RNP and Ca2+ ions with varying concentrations. An additional single-molecule FRET measurement with doubly labelled target DNA revealed that the disappearance of Cy5 signal indicates the dissociation of Cas12a RNP from target DNA without the cleavage reaction (Fig. S5, ESI†). The corresponding dwell time histograms for the R-loop state are shown in the panel to the right of the time traces. (B) Average dwell times of R-loop complex at varying Ca2+ ion concentrations. The average dwell times were obtained by fitting the dwell time histograms in (a) to a single-exponential decay function (red line). | ||
If the divalent metal ions that first participated in the reaction step of the R-loop formation were preserved in the Cas12a R-loop complex, the dwell times of the R-loop complex would not show any appreciable change by increasing the Ca2+ concentration. Therefore, based on our observation regarding the significant positive correlation between the dwell time and the Ca2+ concentration, we speculated that the divalent metal ions are highly mobile while allosterically stabilising the R-loop complex. Consistently, in the experiments with the mixtures with different molar ratios of Mg2+ and Ca2+ ions, we also observed that the number of cleaved DNA substrates proportionally increased by increasing the ratio of Mg2+/Ca2+ concentrations, showing the competitive relation between these two metal ions (Fig. S7, ESI†).
To test this hypothesis, we designed a real-time buffer exchange experiment in which the Ca2+ buffer in the detection chamber was exchanged with a reaction buffer containing catalytically active Mg2+ ions to control the DNA cleavage activity of Cas12a RNP. Surprisingly, in this assay, the Cas12a ternary complex arrested in the pre-cleavage R-loop state in the presence of Ca2+ ions proceeded to the subsequent reaction steps accompanying the final DNA cleavage and release after exchange with the Mg2+ buffer (Fig. 3A). The complete recovery of the DNA cleavage activity indicated that the coordinated Ca2+ ions were successfully replaced with Mg2+ ions. This conclusion was further supported by the observation that, when the Ca2+ buffer in the detection chamber was replaced with a buffer that contained no divalent cations to remove the free Ca2+ ions, the Cas12a RNP was immediately dissociated from the stable R-loop complex (Fig. S8, ESI†). Consistent with the above observations, when the Mg2+ buffer in the detection chamber was replaced with Ca2+ buffer, the cleavage reaction was reversely interrupted (Fig. S9 and S10, ESI†). Furthermore, the use of real-time buffer exchange experiments for the NTS and TS pre-nicked DNA constructs enabled us to independently observe that, in both the NTS and TS pre-cleavage states arrested by Ca2+ ions, the metal ions are exchangeable and thereby the nuclease activity is recovered (Fig. 3B and C). In addition, similar results were obtained in the other Cas12a orthologue (Fig. S11, ESI†). Therefore, we concluded that the divalent metal ions involved in the Cas12a-mediated DNA cleavage reaction are dynamically coordinated in the catalytic site while stabilising the NTS or TS pre-cleavage states.
 | ||
| Fig. 3 Exchangeability of divalent metal ions in the Cas12a-mediated DNA cleavage reaction. Representative time traces observed in real-time buffer exchange experiments for (A) wild-type, (B) TS pre-nicked, and (C) NTS pre-nicked target DNA constructs. A reaction buffer containing 10 mM Mg2+ ions was introduced to the detection chamber in which Cas12a RNP–DNA ternary complexes were arrested in the pre-cleavage state under the reaction condition including 10 mM Ca2+ ions. | ||
A recent crystal structure of Cas12i2 showed that the architecture of the catalytic pocket of Cas12i2 to which the divalent cation is coordinated is very similar to that of RNase H which consists of negatively charged residues that interact with two divalent cations.9 Based on this structural similarity, the complete loss of the endonuclease activity of Cas12a in the presence of Ca2+ ions may be attributed to the relatively long interatomic distance between the two metal ions coordinated in the catalytic pocket, as conjectured by extensive studies on RNase H.20–23 This is because the relatively long interatomic distance between the two coordinated metal ions hampers deprotonation of nucleophile which is an essential for DNA hydrolysis.
Previous studies on the Cas12a-mediated DNA cleavage reaction revealed that a single catalytic site of Cas12a cleaves two DNA strands sequentially.7,8 Hence, the core region containing the catalytic residues that interact with the metal ions and the scissile phosphates of the DNA target would be required to undergo structural rearrangements to enable the two consecutive catalysis of DNA cleavage to occur.24 With this context in mind, the observed dynamic coordination of the metal ions could be advantageous in that they would allow immediate structural rearrangements to support the multiple catalysis of Cas12a. Consistently, previous studies on the metal ion-dependent enzymes such as DNA polymerases and exonucleases, which promote multiple catalytic reactions, also suggested that the dynamic behaviour of metal ions coordinated to the active site of the enzymes may facilitate multiple catalytic turnovers.25,26
In summary, we presented here that the divalent metal ions involved in the DNA cleavage by Cas12a RNP play the unexpected structural role of stabilising the Cas12 pre-cleavage complexes for both NTS and TS cleavage. Notably, by using the ion exchange experiment, we also found that the metal ions are dynamically coordinated in the catalytic centre while stabilizing the NTS or TS pre-cleavage states. These findings not only provide mechanistic insight into the divalent cation-mediated DNA nuclease activity of Cas12a, but also suggest a new strategy for the conditional control of Cas12 functions using the ion exchange method (Fig. 4).
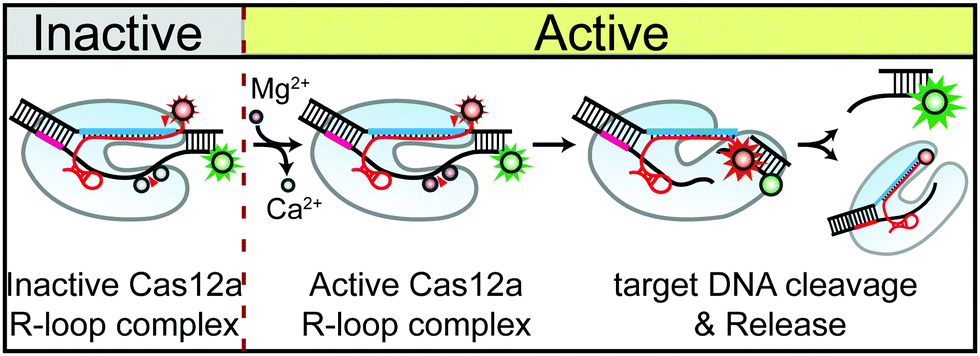 | ||
| Fig. 4 Schematic illustration of conditional control of Cas12a-mediated DNA cleavage using the ion exchange method. | ||
This work was supported by National Research Foundation of Korea (NRF) grants (No. 2020M3A9I4038197 and 2021R1A2C1007593) funded by the Ministry of Science & ICT of the Korean Government, and by the GIST Research Institute (GRI) grant funded by the GIST.
Conflicts of interest
There are no conflicts to declare.Notes and references
- B. Zetsche, M. Heidenreich, P. Mohanraju, I. Fedorova, J. Kneppers, E. M. DeGennaro, N. Winblad, S. R. Choudhury, O. O. Abudayyeh, J. S. Gootenberg, W. Y. Wu, D. A. Scott, K. Severinov, J. van der Oost and F. Zhang, Nat. Biotechnol., 2017, 35, 31–34 CrossRef CAS PubMed.
- J. S. Chen, E. Ma, L. B. Harrington, M. Da Costa, X. Tian, J. M. Palefsky and J. A. Doudna, Science, 2018, 360, 436–439 CrossRef CAS PubMed.
- G. J. Knott and J. A. Doudna, Science, 2018, 361, 866–869 CrossRef CAS PubMed.
- B. P. Kleinstiver, A. A. Sousa, R. T. Walton, Y. E. Tak, J. Y. Hsu, K. Clement, M. M. Welch, J. E. Horng, J. Malagon-Lopez, I. Scarfo, M. V. Maus, L. Pinello, M. J. Aryee and J. K. Joung, Nat. Biotechnol., 2019, 37, 276–282 CrossRef CAS PubMed.
- L. T. Nguyen, B. M. Smith and P. K. Jain, Nat. Commun., 2020, 11, 4906 CrossRef CAS PubMed.
- D. C. Swarts, J. van der Oost and M. Jinek, Mol. Cell, 2017, 66, 221 CrossRef CAS PubMed.
- Y. Jeon, Y. H. Choi, Y. Jang, J. Yu, J. Goo, G. Lee, Y. K. Jeong, S. H. Lee, I. S. Kim, J. S. Kim, C. Jeong, S. Lee and S. Bae, Nat. Commun., 2018, 9, 2777 CrossRef PubMed.
- D. C. Swarts and M. Jinek, Mol. Cell, 2019, 73, 589 CrossRef CAS PubMed.
- X. Huang, W. Sun, Z. Cheng, M. Chen, X. Li, J. Wang, G. Sheng, W. Gong and Y. Wang, Nat. Commun., 2020, 11, 5241 CrossRef CAS PubMed.
- B. Zetsche, J. S. Gootenberg, O. O. Abudayyeh, I. M. Slaymaker, K. S. Makarova, P. Essletzbichler, S. E. Volz, J. Joung, J. van der Oost, A. Regev, E. V. Koonin and F. Zhang, Cell, 2015, 163, 759–771 CrossRef CAS PubMed.
- D. Dong, K. Ren, X. Qiu, J. Zheng, M. Guo, X. Guan, H. Liu, N. Li, B. Zhang, D. Yang, C. Ma, S. Wang, D. Wu, Y. Ma, S. Fan, J. Wang, N. Gao and Z. Huang, Nature, 2016, 532, 522–526 CrossRef CAS PubMed.
- T. Yamano, H. Nishimasu, B. Zetsche, H. Hirano, I. M. Slaymaker, Y. Li, I. Fedorova, T. Nakane, K. S. Makarova, E. V. Koonin, R. Ishitani, F. Zhang and O. Nureki, Cell, 2016, 165, 949–962 CrossRef CAS PubMed.
- B. Li, J. Yan, Y. Zhang, W. Li, C. Zeng, W. Zhao, X. Hou, C. Zhang and Y. Dong, Mol. Ther.--Nucleic Acids, 2020, 19, 1043–1052 CrossRef CAS PubMed.
- D. Singh, J. Mallon, A. Poddar, Y. Wang, R. Tippana, O. Yang, S. Bailey and T. Ha, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 5444–5449 CrossRef CAS PubMed.
- L. Zhang, R. Sun, M. Yang, S. Peng, Y. Cheng and C. Chen, iScience, 2019, 19, 492–503 CrossRef CAS PubMed.
- S. Stella, P. Mesa, J. Thomsen, B. Paul, P. Alcon, S. B. Jensen, B. Saligram, M. E. Moses, N. S. Hatzakis and G. Montoya, Cell, 2018, 175, 1856–1871 e1821 CrossRef CAS PubMed.
- E. Worle, L. Jakob, A. Schmidbauer, G. Zinner and D. Grohmann, Nucleic Acids Res., 2021, 49, 5278–5293 CrossRef PubMed.
- J. C. Cofsky, D. Karandur, C. J. Huang, I. P. Witte, J. Kuriyan and J. A. Doudna, eLife, 2020, 9, e55143 CrossRef CAS PubMed.
- H. Son, J. Park, I. Hwang, Y. Jung, S. Bae and S. Lee, Proc. Natl. Acad. Sci. U. S. A., 2021, 118, e2113747118 CrossRef PubMed.
- M. Nowotny and W. Yang, EMBO J., 2006, 25, 1924–1933 CrossRef CAS PubMed.
- W. Yang, J. Y. Lee and M. Nowotny, Mol. Cell, 2006, 22, 5–13 CrossRef CAS PubMed.
- M. Nowotny, EMBO Rep., 2009, 10, 144–151 CrossRef CAS PubMed.
- E. Rosta, W. Yang and G. Hummer, J. Am. Chem. Soc., 2014, 136, 3137–3144 CrossRef CAS PubMed.
- H. Yang, P. Gao, K. R. Rajashankar and D. J. Patel, Cell, 2016, 167, 1814 CrossRef CAS PubMed.
- J. Gouge, S. Rosario, F. Romain, P. Beguin and M. Delarue, J. Mol. Biol., 2013, 425, 4334–4352 CrossRef CAS PubMed.
- W. Hwang, J. Yoo, Y. Lee, S. Park, P. L. Hoang, H. Cho, J. Yu, T. M. Hoa Vo, M. Shin, M. S. Jin, D. Park, C. Hyeon and G. Lee, Nat. Commun., 2018, 9, 4404 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1cc04446j |
| ‡ Both authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2022 |