
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A novel thiol-labile cysteine protecting group for peptide synthesis based on a pyridazinedione (PD) scaffold†
Richard J.
Spears
 ,
Clíona
McMahon
,
Monika
Shamsabadi
,
Calise
Bahou
,
Ioanna A.
Thanasi
,
Léa N. C.
Rochet
,
Nafsika
Forte
,
Fabien
Thoreau
,
James R.
Baker
* and
Vijay
Chudasama
*
,
Clíona
McMahon
,
Monika
Shamsabadi
,
Calise
Bahou
,
Ioanna A.
Thanasi
,
Léa N. C.
Rochet
,
Nafsika
Forte
,
Fabien
Thoreau
,
James R.
Baker
* and
Vijay
Chudasama
*
UCL Department of Chemistry, 20 Gordon Street, London, WC1H 0AJ, UK. E-mail: v.chudasama@ucl.ac.uk; j.r.baker@ucl.ac.uk
First published on 1st October 2021
Abstract
Herein we report a thiol-labile cysteine protecting group based on an unsaturated pyridazinedione (PD) scaffold. We establish compatibility of the PD in conventional solid phase peptide synthesis (SPPS), showcasing this in the on-resin synthesis of biologically relevant oxytocin. Furthermore, we establish the applicability of the PD protecting group towards both microwave-assisted SPPS and native chemical ligation (NCL) in a model system.
Protecting group strategies for chemoselective protection and deprotection, also referred to as “orthogonal” protecting groups, have proved invaluable in the synthesis of complex biomolecules.1 In particular, construction of peptides containing multiple disulfide bonds frequently relies on orthogonal cysteine protecting groups to engineer the disulfide linkages in a regioselective manner.2 Examples of these include synthesis of hormones such as human insulin3 and bioactive molecules such as conotoxins.4 In addition, orthogonal cysteine protecting groups have also been used in protein semisynthesis5 and more recently in the synthesis of homogeneous antibody drug conjugates (ADCs) bearing multiple payloads.6 The majority of commonly used cysteine protecting groups require acidic conditions and/or toxic heavy metals such as Hg for deprotection, which has thus resulted in a great deal of literature focusing on designing strategies that offer milder deprotection conditions.7,8 In particular, thiol labile protecting groups show promise as orthogonal cysteine protecting groups as these can be removed under mild reducing conditions in peptide synthesis. This has primarily been demonstrated with the tert-butylsulphenyl/tert-butylthio (StBu) protecting group.9 Deprotection of this group is notably sluggish however, requiring deprotection times of several hours or days depending on peptide sequence.10 In an effort to address this, the S-dimethoxyphenylthio/S-trimethoxyphenylthio (S-Dmp/S-Tmp) groups11 and the sec-isoamyl mercaptan/2-methyloxolane-3-thiol (SIT/MOT) groups12 have been reported as replacements for StBu, offering significantly faster deprotection times albeit with minor instability towards strongly acidic conditions/piperidine occasionally observed.11,12 As evidenced by the development of S-Dmp/S-Tmp and SIT/MOT, expansion of the thiol-labile class of cysteine protecting groups would greatly enhance the peptide chemist's current toolkit.
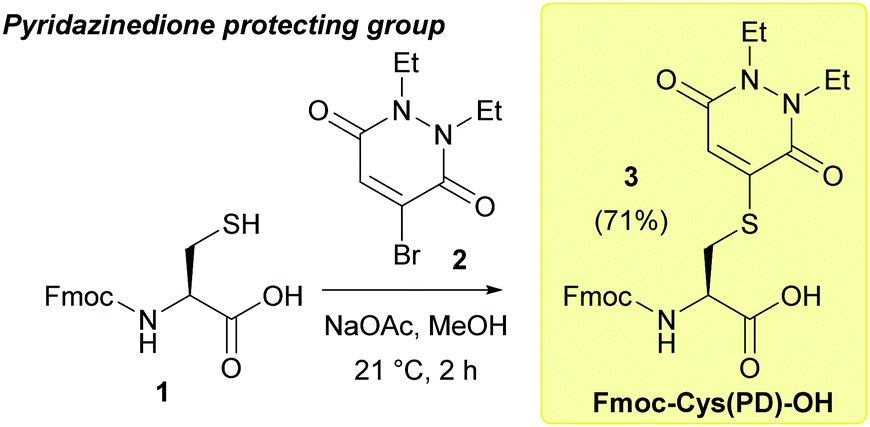 | ||
| Fig. 1 Synthesis of the protected amino acid Fmoc-Cys(PD)-OH 3. | ||
Over the last decade, we have reported pyridazinediones (PDs) as a class of multifunctional molecules for cysteine functionalisation.13 We have found that PDs have proved extremely versatile in organic synthesis and chemical biology14–16 and can be readily cleaved via addition of small molecule thiols in mild, aqueous conditions i.e. phosphate buffer (PB) pH 7–8.13,17 Given these properties, we anticipated the PD would potentially be well suited to act as a novel, thiol-labile cysteine protecting group in peptide synthesis. We began by validating the use of PDs as cysteine protecting groups in conventional 9-fluorenylmethoxycarbonyl solid phase peptide synthesis (Fmoc SPPS). First, we performed a one-step reaction between Fmoc-Cys-OH 1 and bromopyridazinedione 2 to prepare a suitably protected model Fmoc-Cys(PD)-OH 3 in 71% yield (Fig. 1, see ESI† for full synthesis). Fmoc-Cys(PD)-OH 3 was subsequently incorporated into peptides via standard Fmoc SPPS, using 2-(6-chloro-1-H-benzotriazole-1-yl)-1,1,3,3-tetramethylaminium hexafluorophosphate (HCTU) and N,N-diisopropylethylamine (DIPEA) coupling on a 2-chlorotrityl resin with Fmoc deprotections carried out using 20% piperidine in DMF (5 × 2 min treatments). We envisaged that, upon completion of on-resin synthesis, TFA-mediated cleavage of the peptide from the resin (TFA![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :TIS:H2O, 95:2.5:2.5) would give the corresponding PD protected peptide, whereas the free thiol-containing peptide would be obtained instead if the resin was treated with a suitable thiol-containing cocktail prior to resin cleavage. Initially, we synthesised on-resin the protected 7-mer peptide Cys(PD)-Ser(tBu)-Leu-Tyr(tBu)-Arg(Pbf)-Ala-Gly, which was cleaved with TFA:TIS:H2O (95:2.5:2.5). The corresponding PD protected peptide H-Cys(PD)-Ser-Leu-Tyr-Arg-Ala-Gly-OH (C(PD)SLYRAG) 4 (Fig. 2a) was obtained as judged by liquid-chromatography mass spectrometry (LCMS), suggesting the PD protecting group was stable to both the acidic and basic conditions used during Fmoc SPPS. Furthermore, crude peptide 4 was obtained in >95% purity as judged by high-performance liquid chromatography (HPLC) analysis. Next, we investigated conditions for on-resin deprotection of the PD protecting group to synthesise the corresponding CSLYRAG peptide CSLYRAG 5 (Fig. 2b). We have previously established that the unsaturated PD motif in protein–PD bioconjugates can be cleaved by addition of excess thiols.18 With this in mind, we attempted on-resin PD deprotection with 1,4-dithiothreitol (DTT) due to its capacity to remove thiol-labile cysteine protecting groups and its comparative lack of toxicity/stench compared to other thiols such as β-mercaptoethanol (BME) and 1,2-ethanedithiol (EDT). Taking inspiration from the previously described S-Dmp/S-Tmp protecting groups,11 we initially treated resin-bound peptide (following the final Fmoc deprotection cycle) with a 5% DTT in DMF, 0.1 M N-methylmorpholine (NMM) deprotection solution (3 × 5 min treatments). Following peptide cleavage from the resin, we obtained the CSLYRAG peptide CSLYRAG 5 with minimal deprotection of the PD observed by LCMS (see ESI†). In an effort to increase this conversion, we modified the deprotection solution used to comprise an aqueous component, given that our previous work involving deconjugation of protein–PD bioconjugates has primarily been performed in aqueous buffer systems. We therefore treated portions of resin-bound PD protected peptide (3 × 5 min treatments) with deprotection solutions containing either 5% or 10% DTT w/v with differing ratios of DMF and 5 mM PB pH 8.0, followed by peptide cleavage with TFA:TIS:H2O (95:2.5:2.5, with 5% DTT w/v to ensure the liberated thiols were kept in the reduced form). Improved conversions (as judged by LCMS) to the corresponding peptide 5 were observed, particularly when using a 5% or 10% DTT w/v in DMF:5 mM phosphate buffer (8:2) deprotection solution (3 × 5 min treatments), of which the best results were observed when using 10% DTT w/v. Incorporation of Fmoc-Cys(PD)-OH 3 at an internal position within a peptide sequence was then investigated. In a similar manner to the synthesis of 4 and 5, we synthesised peptides SLYC(PD)RAG 6 (Fig. 2c) and SLYCRAG 7 (Fig. 2d) which were obtained following resin cleavage and confirmed by LCMS, with crude purities of 90% and 75% respectively as judged by HPLC. Additionally, we found that no racemisation from L-Cys to D-Cys had occurred in the synthesis of SLYCRAG 7 when using the Cys(PD) protecting group strategy (see ESI†). As a direct comparison for the lability of PD protecting group vs. the StBu protecting group, we also synthesised SLYC(StBu)RAG 8 in addition to SLYC(PD)RAG 6 and attempted on-resin synthesis of SLYCRAG 7 (Fig. 3a), with Cys deprotection performed using a 10% DTT w/v in DMF:5 mM phosphate buffer (8:2) deprotection solution (3 × 5 min treatments) and resin cleavage achieved using TFA:TIS:H2O (95:2.5:2.5, forgoing 5% DTT w/v addition in the cocktail for both peptides; this was done to avoid unintended StBu deprotection unrelated to prior DTT treatment). Synthesis of SLYC(StBu)RAG 8 proceeded similarly to that of SLYC(PD)RAG 6, whereas attempts to synthesise the deprotected peptide 7 using the Cys(StBu) protecting group strategy gave a mixture of deprotected 7/protected peptide 8 (60% deprotected) as judged by HPLC, as opposed to near quantitative deprotection achieved using the Cys(PD) protecting group strategy (>90% deprotected, Fig. 3b). Overall, the PD protecting group shows faster deprotection times than those reported for the StBu protecting group10 and similar to S-Dmp/S-Tmp11 and SIT/MOT.12
:TIS:H2O, 95:2.5:2.5) would give the corresponding PD protected peptide, whereas the free thiol-containing peptide would be obtained instead if the resin was treated with a suitable thiol-containing cocktail prior to resin cleavage. Initially, we synthesised on-resin the protected 7-mer peptide Cys(PD)-Ser(tBu)-Leu-Tyr(tBu)-Arg(Pbf)-Ala-Gly, which was cleaved with TFA:TIS:H2O (95:2.5:2.5). The corresponding PD protected peptide H-Cys(PD)-Ser-Leu-Tyr-Arg-Ala-Gly-OH (C(PD)SLYRAG) 4 (Fig. 2a) was obtained as judged by liquid-chromatography mass spectrometry (LCMS), suggesting the PD protecting group was stable to both the acidic and basic conditions used during Fmoc SPPS. Furthermore, crude peptide 4 was obtained in >95% purity as judged by high-performance liquid chromatography (HPLC) analysis. Next, we investigated conditions for on-resin deprotection of the PD protecting group to synthesise the corresponding CSLYRAG peptide CSLYRAG 5 (Fig. 2b). We have previously established that the unsaturated PD motif in protein–PD bioconjugates can be cleaved by addition of excess thiols.18 With this in mind, we attempted on-resin PD deprotection with 1,4-dithiothreitol (DTT) due to its capacity to remove thiol-labile cysteine protecting groups and its comparative lack of toxicity/stench compared to other thiols such as β-mercaptoethanol (BME) and 1,2-ethanedithiol (EDT). Taking inspiration from the previously described S-Dmp/S-Tmp protecting groups,11 we initially treated resin-bound peptide (following the final Fmoc deprotection cycle) with a 5% DTT in DMF, 0.1 M N-methylmorpholine (NMM) deprotection solution (3 × 5 min treatments). Following peptide cleavage from the resin, we obtained the CSLYRAG peptide CSLYRAG 5 with minimal deprotection of the PD observed by LCMS (see ESI†). In an effort to increase this conversion, we modified the deprotection solution used to comprise an aqueous component, given that our previous work involving deconjugation of protein–PD bioconjugates has primarily been performed in aqueous buffer systems. We therefore treated portions of resin-bound PD protected peptide (3 × 5 min treatments) with deprotection solutions containing either 5% or 10% DTT w/v with differing ratios of DMF and 5 mM PB pH 8.0, followed by peptide cleavage with TFA:TIS:H2O (95:2.5:2.5, with 5% DTT w/v to ensure the liberated thiols were kept in the reduced form). Improved conversions (as judged by LCMS) to the corresponding peptide 5 were observed, particularly when using a 5% or 10% DTT w/v in DMF:5 mM phosphate buffer (8:2) deprotection solution (3 × 5 min treatments), of which the best results were observed when using 10% DTT w/v. Incorporation of Fmoc-Cys(PD)-OH 3 at an internal position within a peptide sequence was then investigated. In a similar manner to the synthesis of 4 and 5, we synthesised peptides SLYC(PD)RAG 6 (Fig. 2c) and SLYCRAG 7 (Fig. 2d) which were obtained following resin cleavage and confirmed by LCMS, with crude purities of 90% and 75% respectively as judged by HPLC. Additionally, we found that no racemisation from L-Cys to D-Cys had occurred in the synthesis of SLYCRAG 7 when using the Cys(PD) protecting group strategy (see ESI†). As a direct comparison for the lability of PD protecting group vs. the StBu protecting group, we also synthesised SLYC(StBu)RAG 8 in addition to SLYC(PD)RAG 6 and attempted on-resin synthesis of SLYCRAG 7 (Fig. 3a), with Cys deprotection performed using a 10% DTT w/v in DMF:5 mM phosphate buffer (8:2) deprotection solution (3 × 5 min treatments) and resin cleavage achieved using TFA:TIS:H2O (95:2.5:2.5, forgoing 5% DTT w/v addition in the cocktail for both peptides; this was done to avoid unintended StBu deprotection unrelated to prior DTT treatment). Synthesis of SLYC(StBu)RAG 8 proceeded similarly to that of SLYC(PD)RAG 6, whereas attempts to synthesise the deprotected peptide 7 using the Cys(StBu) protecting group strategy gave a mixture of deprotected 7/protected peptide 8 (60% deprotected) as judged by HPLC, as opposed to near quantitative deprotection achieved using the Cys(PD) protecting group strategy (>90% deprotected, Fig. 3b). Overall, the PD protecting group shows faster deprotection times than those reported for the StBu protecting group10 and similar to S-Dmp/S-Tmp11 and SIT/MOT.12
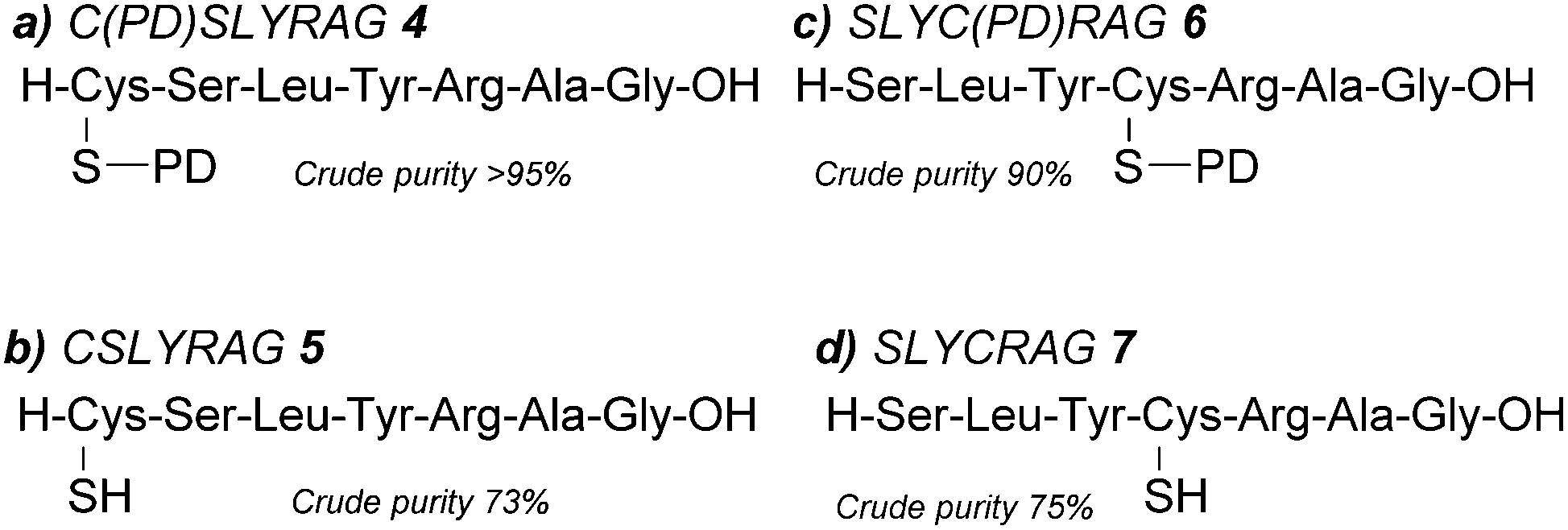 | ||
| Fig. 2 Peptides synthesised (a) C(PD)SLYRAG 4, (b) CSLYRAG 5, (c) SLYC(PD)RAG 6 and (d) SLYCRAG 7. | ||
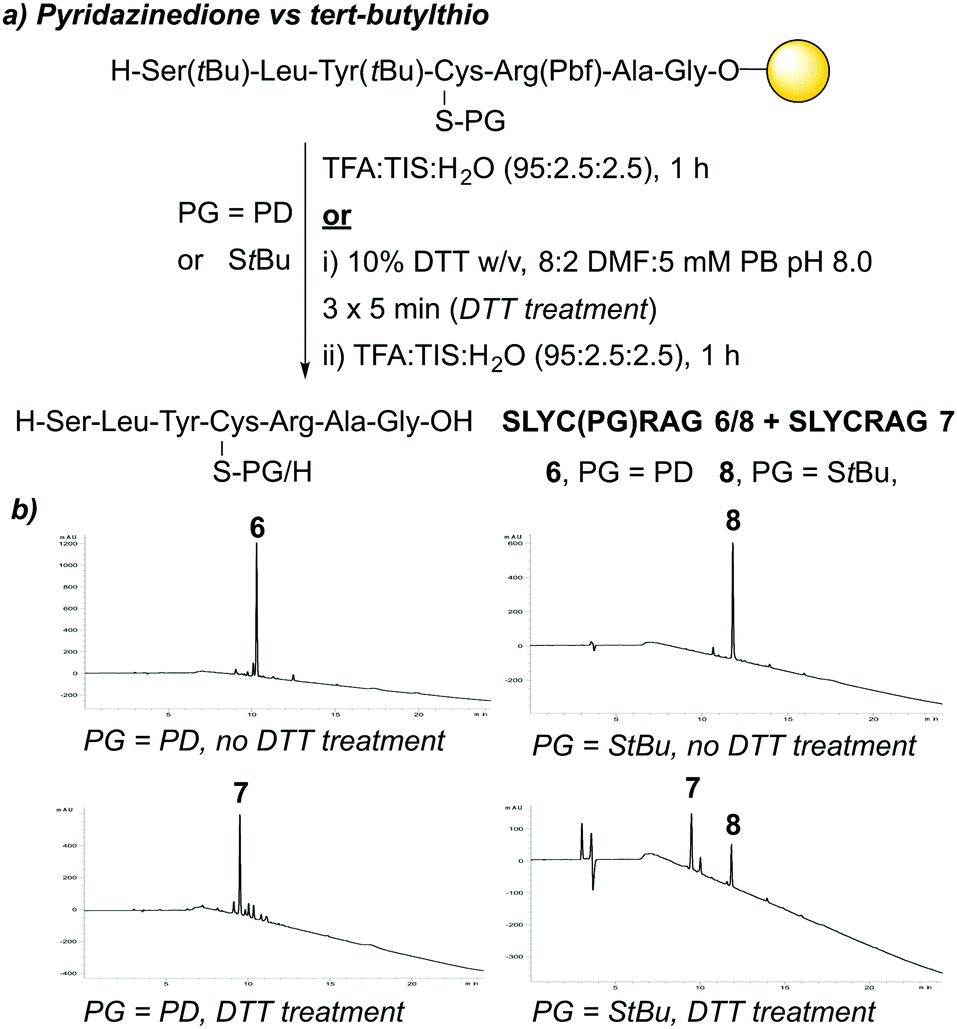 | ||
| Fig. 3 (a) Outline of synthesis of peptides 6, 7, and 8. (b) HPLC data obtained for attempted synthesis of peptides 6 and 8, along with attempted synthesis of 7. | ||
Previously published work featuring PDs reports that the unsaturated motif typically displays an absorption maximum at ca. λ = 300–350 nm, e.g. ε345 = 9100 cm−1 for a dithiopyridazinedione.19 We hypothesised that on-resin PD deprotection would yield thiol–PD adducts that could be monitored by performing UV-vis analysis of wash solutions post incubation with a given resin-bound, PD protected peptide (Fig. 4a). To test this hypothesis, we collected the wash solutions from resin-bound peptides that had been treated with our deprotection solutions and performed UV-vis analysis. We observed a large absorbance readout that decreased with each washing of the resin; for example, in the synthesis of SLYCRAG 7 (Fig. 4b). This decrease in absorbance was also found to coincide with efficient PD deprotection as judged by LCMS (following resin cleavage). We found that UV-vis analysis of wash solutions collected after treating the resin with deprotection solution in this manner proved to be a rapid and reliable way of tracking PD deprotection during peptide synthesis without the need to cleave portions of the resin-bound peptide to analyse deprotection by HPLC or LCMS. We therefore continued to use this method of analysis throughout the remainder of our studies of the PD protecting group.
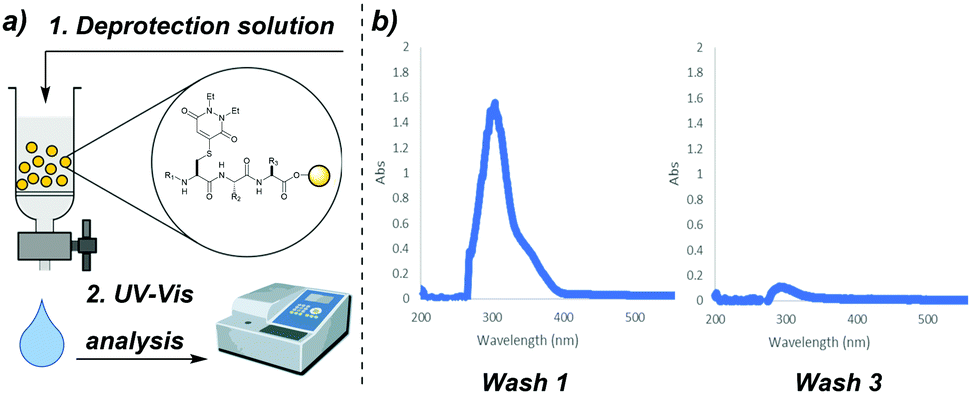 | ||
| Fig. 4 (a) Outline monitoring on-resin PD deprotection by UV-vis analysis of wash solutions. (b) Example UV-vis traces of wash solutions in the synthesis of SLYCRAG 7. | ||
We next investigated the applicability of the PD protecting group towards the synthesis of peptides containing more than one cysteine. To this end, we focused on synthesising oxytocin, a disulfide containing 9-mer peptide which can be used for various therapeutic applications such as induction of labour.20 We performed on-resin peptide synthesis on a Rink Amide resin via Fmoc SPPS, with Fmoc-Cys(PD)-OH 3 incorporated at both positions 1 and 6 of the peptide. Following the final coupling/Fmoc deprotection step and subsequent peptide cleavage from the resin with TFA:TIS:H2O (95:2.5:2.5), successful synthesis of the crude PD protected linear oxytocin 9 was confirmed by LCMS analysis and obtained in 87% purity as judged by HPLC. Next, we performed stepwise PD deprotection and subsequent thiol oxidation for the on-resin synthesis of oxytocin. PD deprotection of the resin-bound protected linear oxytocin was performed using a 10% w/v DTT in DMF:5 mM phosphate buffer (8:2) deprotection solution (7 × 5 min treatments, deprotection tracked by UV-vis analysis). Following quantitative PD deprotection (as judged by HPLC/UV-vis analysis), on-resin oxidation of the resin-bound, protected, free thiol-containing linear oxytocin peptide was carried out using N-chlorosuccinimide (NCS) as reported previously by Postma and Albericio.21 Following thiol oxidation and peptide cleavage from the resin with TFA:TIS:H2O (95:2.5:2.5), the crude material was subsequently purified by semi-preparative HPLC, with purified oxytocin 10 confirmed by LCMS (Fig. 5a–d).
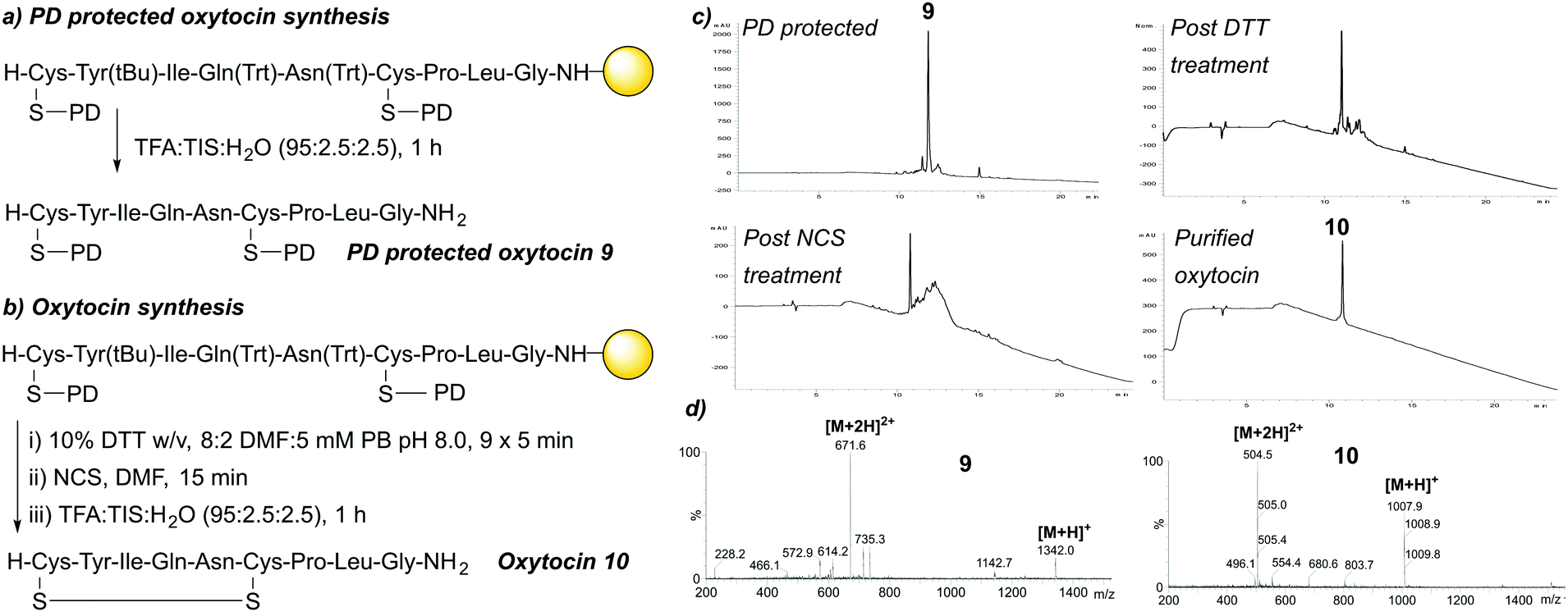 | ||
| Fig. 5 (a) SPPS of protected linear oxytocin 9. (b) SPPS of oxytocin 10. (c) HPLC data for the synthesis of oxytocin 10. (d) LCMS data for 9 and purified 10. | ||
Finally, we sought to investigate the applicability of the PD protecting group within two different aspects of peptide synthesis as a proof of concept. We first focused on the compatibility of the PD protecting group with microwave-assisted peptide synthesis. We synthesised SLYC(PD)RAG 6 on-resin using microwave-assisted conditions (see ESI† for details) for Fmoc-amino acid couplings (Fig. 6a). As a test of stability, upon completion of on-resin synthesis we also subjected the resin-bound peptide to 20% piperidine in DMF under microwave-assisted conditions. The desired PD protected peptide was obtained in a crude purity of 82% (Fig. 6b), suggesting the PD protecting group was compatible with microwave-assisted conditions. Next, we turned our attention to native chemical ligation (NCL),22 a well-documented strategy for the synthesis of large peptides/proteins through reaction of an N-terminal cysteine-containing peptide fragment and a C-terminal thioester-containing fragment.23 We envisaged that addition of thiol would act to deprotect the PD protecting group to yield the N-terminal cysteine required for NCL and that this could be done in a one pot manner along with NCL in the presence of a suitable thioester fragment. We attempted a one-pot PD deprotection/NCL procedure by incubating C(PD)SLYRAG 4 with a model N-acetylglycine thioester 11 in the presence of either DTT or 4-mercaptophenylacetic acid (MPAA) in ammonium acetate (NH4OAc) pH 6.8 for 18 h at 37 °C (Fig. 6c). Total consumption of the PD protected peptide 4 was observed, along with conversion to the anticipated NCL product Ac-GCSLYRAG-COOH 12 as judged by LCMS analysis (Fig. 6d).
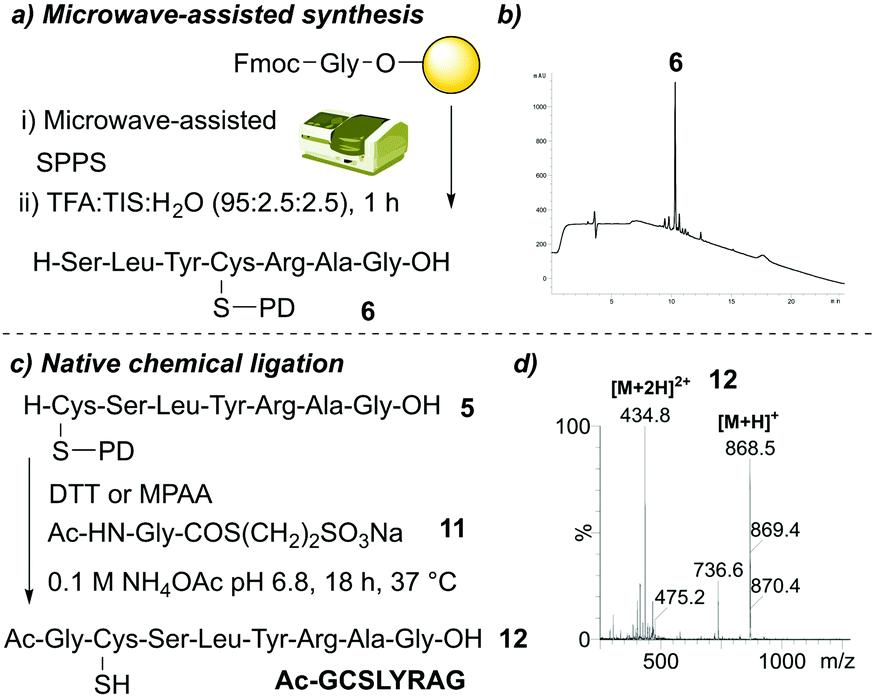 | ||
| Fig. 6 (a) Outline of microwave-assisted synthesis of SLYC(PD)RAG 6. (b) HPLC data obtained. (c) Outline of Ac-GCSLYRAG 12 synthesis. (d) LCMS data of crude reaction containing peaks corresponding to 12. | ||
In conclusion, we have explored the use of pyridazinediones in peptide synthesis, establishing the motif as a thiol-labile protecting group compatible with Fmoc SPPS. We found on-resin deprotection of the PD could be achieved through addition of thiols and monitored by UV-vis analysis. We have demonstrated the applicability of the PD protecting group within on-resin synthesis of disulfide-containing oxytocin, microwave-assisted synthesis and peptide coupling via a one pot deprotection/NCL procedure under aqueous conditions. We anticipate the ability to track thiol deprotection by UV-vis analysis, as well as the potential to diversify the ethyl chains of the PD scaffold with moieties such as affinity/solubility tags and clickable handles will assist in enabling more complex peptide syntheses. Furthermore, given its compatibility with aqueous conditions, we anticipate the PD protecting group could be applicable within research focused on designing more environmentally-friendly, “greener” peptide synthesis.24
We gratefully acknowledge the Leverhulme Trust (RPG-2017-288, 176274 and RPG-2020-010) for funding R. J. S. and C. J. B. (respectively), the EU (projects 859458 and 838703 respectively) for funding L. N. C. R. and F. T. (respectively), and the EPSRC (EP/T517793/1 and EP/R034621/1) for funding I. A. T. and N. F. (respectively). We also acknowledge the UCL Chemistry Mass Spectrometry (MS) Facility (Dr Kersti Karu).
Conflicts of interest
V. C. and J. R. B. are Directors of the spin-out ThioLogics, but there are no competing financial interests to declare.References
- A. Isidro-Llobet, M. Álvarez and F. Albericio, Chem. Rev., 2009, 109, 2455–2504 CrossRef CAS PubMed.
- R. He, J. Pan, J. P. Mayer and F. Liu, ChemBioChem, 2020, 21, 1101–1111 CrossRef CAS PubMed.
- K. Akaji, K. Fujino, T. Tatsumi and Y. Kiso, J. Am. Chem. Soc., 1993, 115, 11384–11392 CrossRef CAS.
- A. Cuthbertson and B. Indrevoll, Org. Lett., 2003, 5, 2955–2957 CrossRef CAS PubMed.
- M. Jbara, S. K. Maity, M. Seenaiah and A. Brik, J. Am. Chem. Soc., 2016, 138, 5069–5075 CrossRef CAS PubMed.
- M. R. Levengood, X. Zhang, J. H. Hunter, K. K. Emmerton, J. B. Miyamoto, T. S. Lewis and P. D. Senter, Angew. Chem., Int. Ed., 2017, 56, 733–737 CrossRef CAS PubMed.
- R. J. Spears, C. McMahon and V. Chudasama, Chem. Soc. Rev., 2021, 50, 11098–11155 RSC.
- S. Laps, G. Satish and A. Brik, Chem. Soc. Rev., 2021, 50, 2367–2387 RSC.
- R. Eritja, J. P. Ziehler-Martin, P. A. Walker, T. D. Lee, K. Legesse, F. Albericio and B. E. Kaplan, Tetrahedron, 1987, 43, 2675–2680 CrossRef CAS.
- M. Góngora-Benítez, J. Tulla-Puche, M. Paradís-Bas, O. Werbitzky, M. Giraud and F. Albericio, Biopolymers, 2011, 96, 69–80 CrossRef PubMed.
- T. M. Postma, M. Giraud and F. Albericio, Org. Lett., 2012, 14, 5468–5471 CrossRef CAS PubMed.
- A. Chakraborty, A. Sharma, F. Albericio and B. G. de la Torre, Org. Lett., 2020, 22, 9644–9647 CrossRef CAS PubMed.
- V. Chudasama, M. E. B. Smith, F. F. Schumacher, D. Papaioannou, G. Waksman, J. R. Baker and S. Caddick, Chem. Commun., 2011, 47, 8781–8783 RSC.
- J. C. F. Nogueira, K. Paliashvili, A. Bradford, F. Di Maggio, D. A. Richards, R. M. Day and V. Chudasama, Org. Biomol. Chem., 2020, 18, 2215–2218 RSC.
- C. Bahou, R. J. Spears, A. E. Aliev, A. Maruani, M. Fernandez, F. Javaid, P. A. Szijj, J. R. Baker and V. Chudasama, Chem. Commun., 2019, 55, 14829–14832 RSC.
- A. Maruani, P. A. Szijj, C. Bahou, J. C. F. Nogueira, S. Caddick, J. R. Baker and V. Chudasama, Bioconjugate Chem., 2020, 31, 520–529 CrossRef CAS PubMed.
- M. Fernández, A. Shamsabadi and V. Chudasama, Chem. Commun., 2020, 56, 1125–1128 RSC.
- C. Bahou, P. A. Szijj, R. J. Spears, A. Wall, F. Javaid, A. Sattikar, E. A. Love, J. R. Baker and V. Chudasama, Bioconjugate Chem., 2021, 32, 672–679 CrossRef CAS PubMed.
- A. Maruani, M. E. B. Smith, E. Miranda, K. A. Chester, V. Chudasama and S. Caddick, Nat. Commun., 2015, 6, 6645 CrossRef CAS PubMed.
- J. M. Shyken and R. H. Petrie, Clin. Obstet. Gynecol., 1995, 38, 232–245 CrossRef CAS PubMed.
- T. M. Postma and F. Albericio, Org. Lett., 2013, 15, 616–619 CrossRef CAS PubMed.
- P. E. Dawson, T. W. Muir, I. Clark-Lewis and S. B. Kent, Science, 1994, 266, 776–779 CrossRef CAS PubMed.
- V. Agouridas, O. El Mahdi, V. Diemer, M. Cargoët, J.-C. M. Monbaliu and O. Melnyk, Chem. Rev., 2019, 119, 7328–7443 CrossRef CAS PubMed.
- O. Al Musaimi, B. G. de la Torre and F. Albericio, Green Chem., 2020, 22, 996–1018 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1cc03802h |
| This journal is © The Royal Society of Chemistry 2022 |