
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
In situ silane activation enables catalytic reduction of carboxylic acids†
Emma L.
Stoll
 a,
Thomas
Barber
a,
David J.
Hirst
b and
Ross M.
Denton
*a
a,
Thomas
Barber
a,
David J.
Hirst
b and
Ross M.
Denton
*a
aSchool of Chemistry, University of Nottingham, GlaxoSmithKline Carbon Neutral Laboratories for Sustainable Chemistry, 6 Triumph Road, Nottingham, NG7 2GA, UK. E-mail: ross.denton@nottingham.ac.uk
bGlaxoSmithKline, Gunnels Wood Road, Stevenage, SG1 2NY, UK
First published on 20th January 2022
Abstract
We describe a catalytic system for the conversion of carboxylic acids into alcohols using substoichiometric zinc acetate and N-methyl morpholine, in combination with phenylsilane as the nominal terminal reductant. Reaction monitoring by 19F NMR spectroscopy demonstrates that the reaction proceeds by mutual activation of the carboxylic acid and silane through the in situ generation of silyl ester intermediates.
The reduction of carboxylic acids to alcohols is an important transformation in organic synthesis, particularly as carboxylic acids are abundant starting materials.1–4 Classically, reductions of this type are carried out using either stoichiometric aluminium reagents such as LiAlH4,5 DIBAL6 and AlH37–9 or borane adducts.10,11 While these reagents are widely used their high reactivity renders many of them air and moisture sensitive and their multi-hydridic nature also generates hazards when quenching reactions. Converting carboxylic acids to activated derivatives, such as benzotriazoles,12–15 boronate esters16,17 or mixed anhydrides18–21 allows reduction to alcohols under much milder conditions, allowing greater functional group tolerance but this is achieved at the expense of atom efficiency as stoichiometric activating agents are required.22 A more atom economical approach involves catalytic hydrogenation using hydrogen gas,23–26 however these reactions require harsh reaction conditions such as high temperatures and pressures. Silane and pinacol borane-mediated16 reductions represent an attractive alternative to the methods above as these reductants are readily available and can be activated by a wide variety of metal catalysts. To date a number of such metal/silane reductions have been reported including Fe,27 Cu,28 In,29 Ru,30,31 and Mn32,33 systems. However, a catalytic hydrosilylation of carboxylic acids that has wide substrate scope and can be implemented without rigorous exclusion of air and moisture or recourse to Schlenk apparatus remains to be developed.
In seeking a practical approach to carboxylic acid reduction, we reasoned that in situ modification could be exploited to activate both the carboxylic acid substrate and the silane reductant through the generation of silyl esters (Fig. 1). This reaction design was predicated on previous observations that silyl esters function as activated carboxylic acids and that they are more potent reductants than phenylsilane.34,35 Herein, we demonstrate that phenylsilane, in combination with substoichiometric N-methylmorpholine and Zn(OAc)2, effects the reduction of a range of carboxylic acids. This gives rise to a practical method for the reduction of carboxylic acids in standard laboratory glassware and demonstrates the principle of in situ silane activation of substrate and reductant.
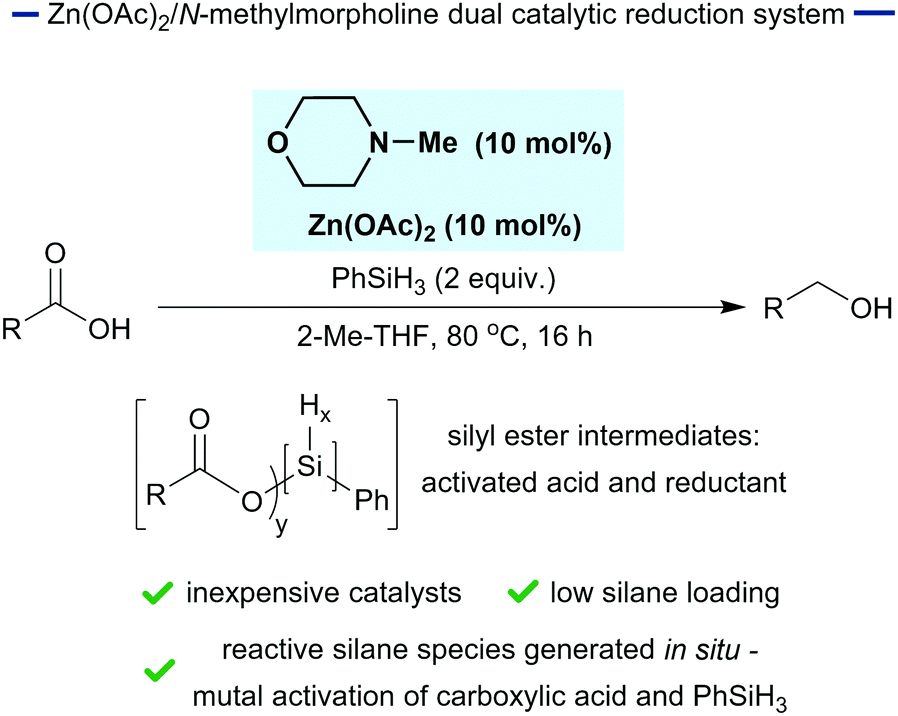 | ||
| Fig. 1 Catalytic reduction of carboxylic acids. | ||
We began by establishing that a combination of Zn(OAc)2 and phenylsilane was active for the reduction of para-fluorobenzoic acid.36 This combination has been used effectively for the hydrosilylation of amides37–40 and we were pleased to observe a moderate 53% of the corresponding alcohol (entry 1, Table 1).
We next explored the addition of N-methylmorpholine which we expected would catalyse a dehydrogenative silylation reaction between the carboxylic acid and silane generating the silyl ester intermediates that we sought.41 Pleasingly, this resulted in a significant increase in yield (entries 2, 3 and 4, Table 1) up to 99% in the case of entry 4 where 20 mol% of N-methylmorpholine was used. However, the reaction was also very efficient when 10 mol% of N-methylmorpholine was used giving an 87% yield of the alcohol (entry 3).
The reduction process was also sensitive to the stoichiometry of Zn(OAc)2 and a decrease in loading was met with a decrease in yield (entry 5, Table 1). The reaction time could be reduced to 6 hours at the expense of additional N-methylmorpholine (entry 6) and, under the general conditions defined within Table 1, both Zn(OAc)2 and N-methylmorpholine were required for high conversion (entries 1, 7 and 8). The conditions depicted in entry 3 were selected as optimal as they represented a balance between reaction rate and N-methylmorpholine stoichiometry. To assess the scope of this process a range of twenty three carboxylic acids were subjected to the reaction conditions (Table 2) beginning with substituted benzoic acids.
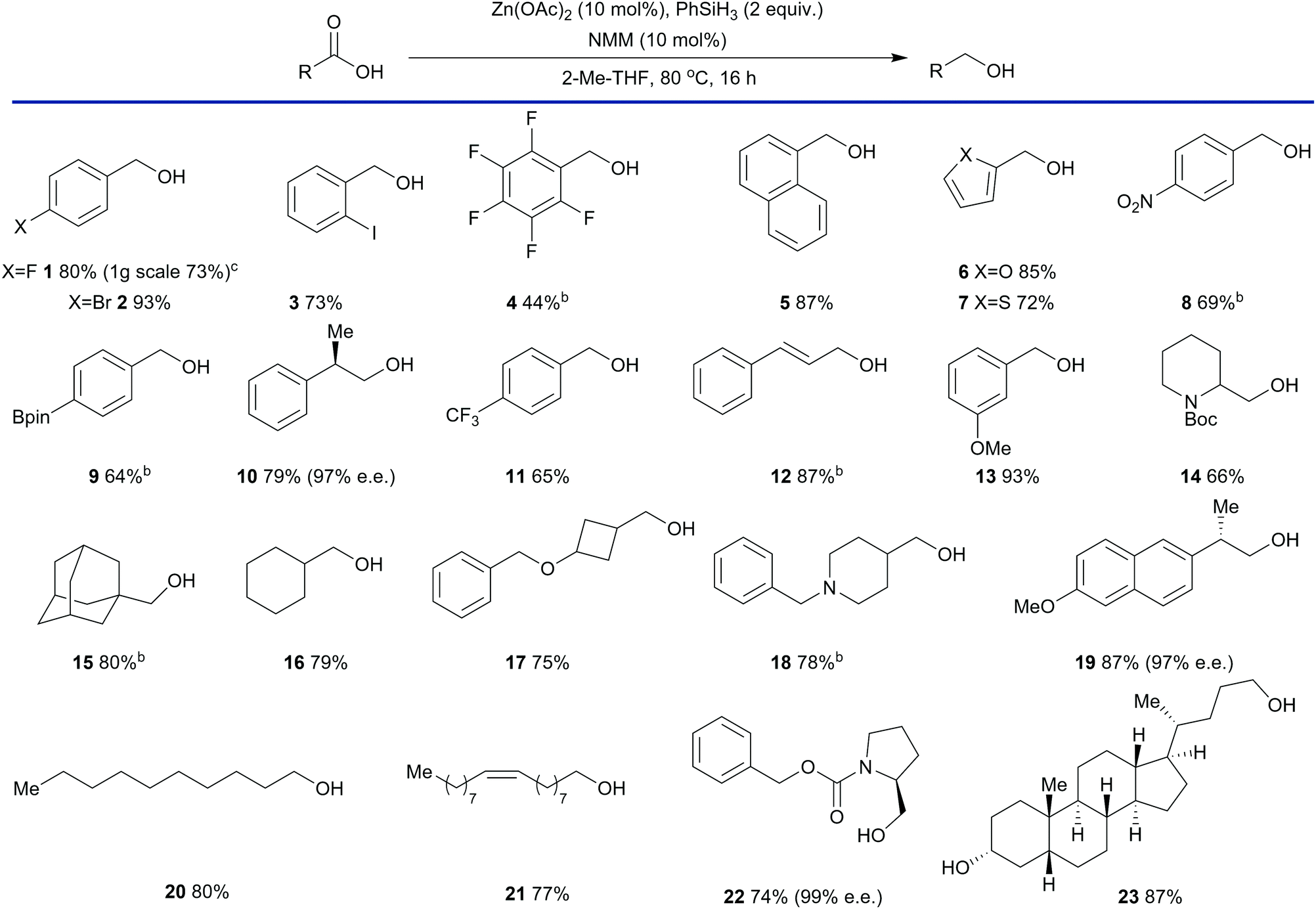
These substrates were reduced in moderate to good yields. Of note are products 2, 3, 8, and 12 which contain potentially reductively labile carbon–halogen bonds, a nitro group and a conjugated alkene respectively. Other aromatic carboxylic, acids such as naphthoic acid (entry 5), 2-furancarboxylic acid (entry 6) and 2-thiophenecarboxylic acid (entry 7), were also reduced in good yields. Similarly, alcohols derived from aliphatic carboxylic acids were also obtained in good yields with both cyclic (entries 14–18) and linear (entries 19–21) substrates well tolerated. Hindered substrates such as adamantane carboxylic acid (entry 15) and substrates containing strained rings such as 3-(benzyloxy)cyclobutyl methanol (entry 17) were also successfully reduced in good yield. A variety of protecting groups were also well tolerated such as carbamates (entries 14 and 22) and benzyl ethers (entry 17). Chiral non-racemic carboxylic acids were also be reduced with very little loss in enantiomeric excess. For example, 10 was obtained with an e.e. of 97%. The naproxen-derived alcohol 19 was obtained with an e.e. of 97% and Cbz-L-prolinol 23 was obtained in 99% e.e. Finally, lithocholic acid (entry 23) was reduced efficiently demonstrating that free hydroxyl groups are not detrimental to the reduction process. Our standard protocol involves carrying out reactions in round bottom flasks fitted with a reflux condenser under a nitrogen or argon atmosphere in anhydrous solvent. However, a 1 g scale reduction was performed using 4-fluorobenzoic acid (entry 1) in Winchester grade 2-Me THF with the reflux condenser open to the air. This resulted in a small reduction in yield from 80% to 73% demonstrating the practicality of the reduction process. Given that several substrates were not soluble in refluxing 2-Me THF products 4, 8, 9, 12, 15 and 18 were obtained by changing the solvent to toluene. Carboxylic acids that are strong Brønsted acids were found to be poor substrates. For example, pentafluorobenzoic acid (entry 4) were gave only 44% yield of the alcohol product. Other problematic substrates including carboxylic acids containing ester, nitrile and amide functional groups (not depicted), which produced a mixture of products.36
Some preliminary investigations were performed to gain insight into the mechanism of the reaction and in particular the proposed generation of silyl ester intermediates (Scheme 1) and reaction monitoring was carried out using fluorobenzoic acid 24 (19F δ = −105.1 ppm). As shown in Scheme 1A and B, in the presence of N-methylmorpholine, formation of silyl esters 25 (19F δ = −104.5 to −106.2 ppm) was observed after one hour. Over the course of the reaction, a steady reduction of the silyl esters was accompanied by an increase in silyl ether peaks 26 (19F δ = −114.9 to −116.0 ppm). The speciation of the silyl esters and ethers is complex as expected because of the trihydridic nature of phenylsilane. However, the silyl ester derived from para-fluorobenzoic acid and chlorophenylsilane (not depicted) was prepared to aid 19F NMR spectroscopy assignment.36
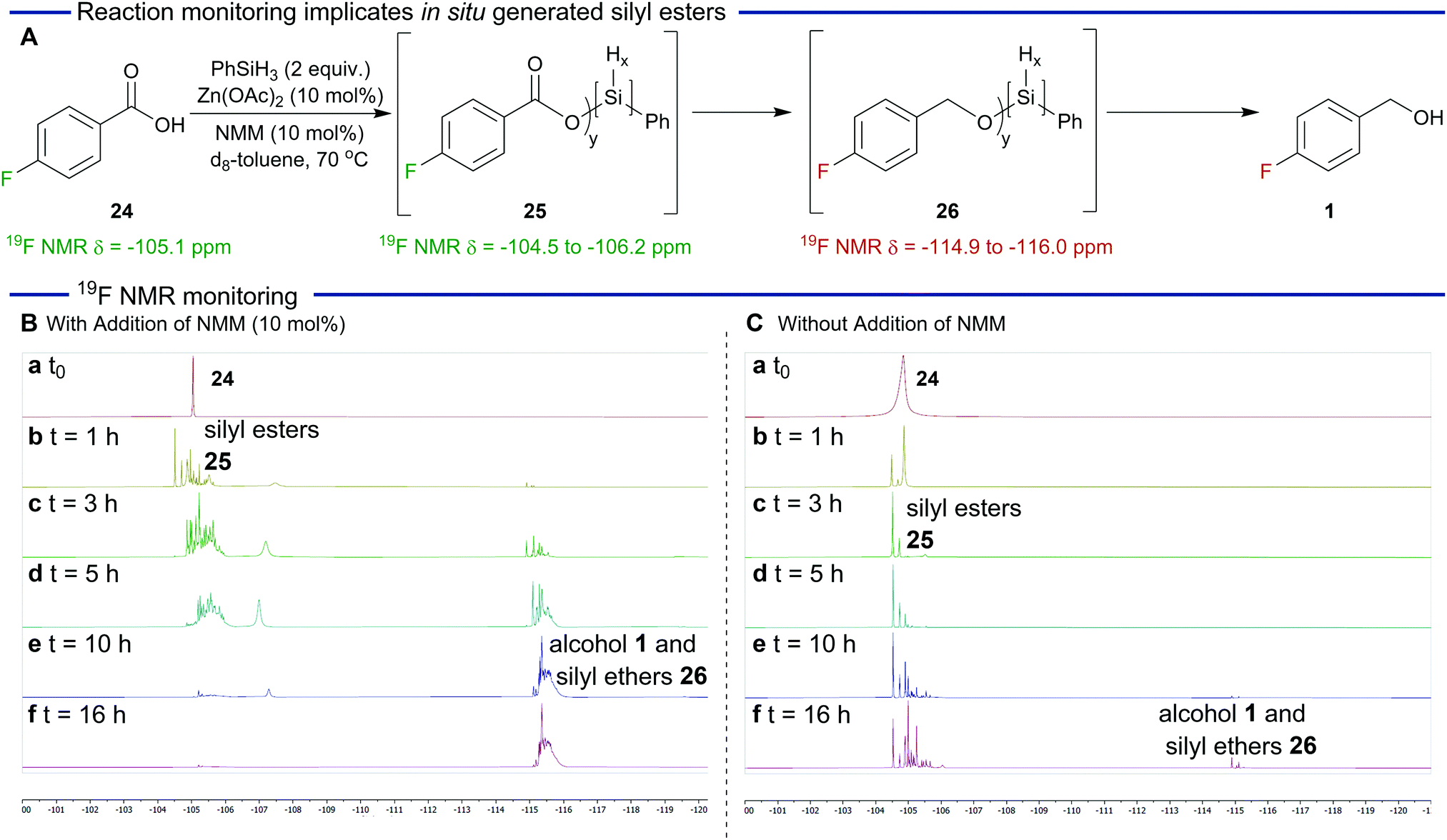 | ||
| Scheme 1 Insights into the mechanism of the reaction. | ||
We next carried out an analogous reduction in the absence of N-methylmorpholine (Scheme 1C). In this case the formation of the silyl esters was significantly slower confirming the key role that N-methylmorpholine plays in their generation. The reduction of these intermediates was also slower and after 16 h silyl esters were still the predominate species in the reaction mixture. This is in sharp contrast with Scheme 1B which shows that the reduction is essentially complete after this time.
In conclusion, we have developed a reduction of carboxylic acids using inexpensive and readily available substoichiometric Zn(OAc)2 and N-methylmorpholine. The reaction can be carried out in conventional glassware and does not require strict exclusion of moisture or air. Reaction monitoring by 19F NMR spectroscopy demonstrates that the reaction proceeds by a mutual activation of the acid and silane through the formation of silylester intermediates. The elucidation of the key role played by these silyl esters and their in situ generation can now be applied to the design of further reactions based on the principle of mutual activation.
Conflicts of interest
There are no conflicts to declare.Notes and references
- J. T. Edwards, R. R. Merchant, K. S. McClymont, K. W. Knouse, T. Qin, L. R. Malins, B. Vokits, S. A. Shaw, D. H. Bao, F. L. Wei, T. Zhou, M. D. Eastgate and P. S. Baran, Nature, 2017, 545, 213–218 CrossRef CAS PubMed.
- C. P. Johnston, R. T. Smith, S. Allmendinger and D. W. C. MacMillan, Nature, 2016, 536, 322–325 CrossRef CAS PubMed.
- A. Fawcett, J. Pradeilles, Y. Wang, T. Mutsuga, E. L. Myers and V. K. Aggarwal, Science, 2017, 357, 283–286 CrossRef CAS PubMed.
- L. J. Gooßen, N. Rodríguez and K. Gooßen, Angew. Chem., Int. Ed., 2008, 47, 3100–3120 CrossRef PubMed.
- R. F. Nystrom and W. G. Brown, J. Am. Chem. Soc., 1947, 69, 2548–2549 CrossRef CAS.
- N. M. Yoon and Y. S. Gyoung, J. Org. Chem., 1985, 50, 2443–2450 CrossRef CAS.
- H. C. Brown and N. M. Yoon, J. Am. Chem. Soc., 1966, 88, 1464–1472 CrossRef CAS.
- N. M. Yoon and H. C. Brown, J. Am. Chem. Soc., 1968, 90, 2927–2938 CrossRef CAS.
- J. R. Hanson, Reductions by the Alumino- and Borohydrides in Organic Synthesis, 2nd edn, 1992 Search PubMed.
- N. M. Yoon, C. S. Pak, H. C. Brown, S. Krishnamurthy and T. P. Stocky, J. Org. Chem., 1973, 38, 2786–2792 CrossRef CAS.
- C. F. Lane, H. L. Myatt, J. Daniels and H. B. Hopps, J. Org. Chem., 1974, 39, 3052–3054 CrossRef CAS.
- J. A. Morales-Serna, E. García-Ríos, J. Bernal, E. Paleo, R. Gaviño and J. Cárdenas, Synthesis, 2011, 1375–1382 CAS.
- K. N. Singh and A. Kaur, Synth. Commun., 2005, 35, 2935–2937 CrossRef.
- R. P. McGeary, Tetrahedron Lett., 1998, 39, 3319–3322 CrossRef CAS.
- T. Okawara, N. Ikeda, T. Yamasaki and M. Furukawa, Chem. Pharm. Bull., 1988, 36, 3628–3631 CrossRef CAS.
- A. Harinath, J. Bhattacharjee and T. K. Panda, Chem. Commun., 2019, 55, 1386–1389 RSC.
- R. H. Tale, K. M. Patil and S. E. Dapurkar, Tetrahedron Lett., 2003, 44, 3427–3428 CrossRef CAS.
- G. Kokotos, Synthesis, 1990, 299–301 CrossRef CAS.
- B. P. Bandgar, R. K. Modhave, P. P. Wadgaonkar and A. R. Sande, J. Chem. Soc., Perkin Trans. 1, 1996, 1993–1994 RSC.
- M. Rodriguez, M. Llinares, S. Doulut, A. Heitz and J. Martinez, Tetrahedron Lett., 1991, 32, 923–926 CrossRef CAS.
- K. Soai, S. Yokoyama and K. Mochida, Synthesis, 1987, 647–648 CrossRef CAS.
- B. M. Trost, Science, 1991, 254, 1471–1477 CrossRef CAS PubMed.
- M. Hudlický, Reductions in Organic Chemistry, John Wiley & Sons, Ltd., 1984 Search PubMed.
- D. H. He, N. Wakasa and T. Fuchikami, Tetrahedron Lett., 1995, 36, 1059–1062 CrossRef CAS.
- J. Ullrich and B. Breit, ACS Catal., 2018, 8, 785–789 CrossRef CAS.
- T. J. Korstanje, J. I. Van Der Vlugt, C. J. Elsevier and B. De Bruin, Science, 2015, 350, 298–302 CrossRef CAS PubMed.
- L. C. M. Castro, H. Li, J. Sortais and C. Darcel, Chem. Commun., 2012, 48, 10514–10516 RSC.
- S. Laval, W. Dayoub, A. Favre-Reguillon, M. Berthod, P. Demonchaux, G. Mignani and M. Lemaire, Tetrahedron Lett., 2009, 50, 7005–7007 CrossRef CAS.
- N. Sakai, K. Kawana, R. Ikeda, Y. Nakaike and T. Konakahara, Eur. J. Org. Chem., 2011, 3178–3183 CrossRef CAS.
- K. Matsubara, T. Iura, T. Maki and H. Nagashima, J. Org. Chem., 2002, 67, 4985–4988 CrossRef CAS PubMed.
- J. A. Fernández-Salas, S. Manzini and S. P. Nolan, Adv. Synth. Catal., 2014, 356, 308–312 CrossRef.
- O. Martínez-Ferraté, B. Chatterjee, C. Werlé and W. Leitner, Catal. Sci. Technol., 2019, 9, 6370–6378 RSC.
- E. Antico, P. Schlichter, C. Werlé and W. Leitner, JACS Au, 2021, 1(6), 742–749 CrossRef CAS PubMed.
- E. L. Stoll, T. Tongue, K. G. Andrews, D. Valette, D. J. Hirst and R. M. Denton, Chem. Sci., 2020, 11, 9494–9500 RSC.
- M. C. D’Amaral, N. Jamkhou and M. J. Adler, Green Chem., 2021, 23, 288–295 RSC.
- For full details see the ESI†.
- S. Das, D. Addis, K. Junge and M. Beller, Chem. – Eur. J., 2011, 17, 12186–12192 CrossRef CAS PubMed.
- S. Das, D. Addis, S. Zhou, K. Junge and M. Beller, J. Am. Chem. Soc., 2010, 132, 1770–1771 CrossRef CAS PubMed.
- K. G. Andrews, D. M. Summers, L. J. Donnelly and R. M. Denton, Chem. Commun., 2016, 52, 1855–1858 RSC.
- C. Cheng and M. Brookhart, J. Am. Chem. Soc., 2012, 134, 11304–11307 CrossRef CAS PubMed.
- K. G. Andrews, R. Faizova and R. M. Denton, Nat. Commun., 2017, 8, 15913 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Details of experimental procedures and characterisation of compounds. See DOI: 10.1039/d1cc03396d |
| This journal is © The Royal Society of Chemistry 2022 |