
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceLysine-targeting inhibition of amyloid β oligomerization by a green perilla-derived metastable chalcone in vitro and in vivo†
Kazuma
Murakami
 *a,
Yoshiki
Sakaguchi
a,
Kota
Taniwa
a,
Naotaka
Izuo‡
b,
Mizuho
Hanaki
a,
Taiji
Kawase
c,
Kenji
Hirose
c,
Takahiko
Shimizu§
b and
Kazuhiro
Irie
*a
*a,
Yoshiki
Sakaguchi
a,
Kota
Taniwa
a,
Naotaka
Izuo‡
b,
Mizuho
Hanaki
a,
Taiji
Kawase
c,
Kenji
Hirose
c,
Takahiko
Shimizu§
b and
Kazuhiro
Irie
*a
aDivision of Food Science and Biotechnology, Graduate School of Agriculture, Kyoto University, Kyoto, 606-8502, Japan. E-mail: murakami.kazuma.4v@kyoto-u.ac.jp; irie.kazuhiro.2z@kyoto-u.ac.jp
bDepartment of Endocrinology, Hematology and Gerontology, Graduate School of Medicine, Chiba University, Chiba, 260-8670, Japan
cNihon Waters, K.K., Tokyo, 140-0001, Japan
First published on 18th October 2022
Abstract
Oligomers of amyloid β (Aβ) represent an early aggregative form that causes neurotoxicity in the pathogenesis of Alzheimer's disease (AD). Thus, preventing Aβ aggregation is important for preventing AD. Despite intensive studies on dietary compounds with anti-aggregation properties, some identified compounds are susceptible to autoxidation and/or hydration upon incubation in water, leaving unanswered issues regarding which active structures in metastable compounds are actually responsible for the inhibition of Aβ aggregation. In this study, we observed the site-specific inhibition of 42-mer Aβ (Aβ42) oligomerization by the green perilla-derived chalcone 2′,3′-dihydroxy-4′,6′-dimethoxychalcone (DDC), which was converted to its decomposed flavonoids (dDDC, 1–3) via nucleophilic aromatic substitution with water molecules. DDC suppressed Aβ42 fibrillization and slowed the transformation of the β-sheet structure, which is rich in Aβ42 aggregates. To validate the contribution of dDDC to the inhibitory effects of DDC on Aβ42 aggregation, we synthesized 1–3 and identified 3, a catechol-type flavonoid, as one of the active forms of DDC. 1H–15N SOFAST-HMQC NMR revealed that 1–3 as well as DDC could interact with residues between His13 and Leu17, which were near the intermolecular β-sheet (Gln15–Ala21). The nucleation in Aβ42 aggregates involves the rate-limiting formation of low-molecular-weight oligomers. The formation of a Schiff base with dDDC at Lys16 and Lys28 in the dimer through autoxidation of dDDC was associated with the suppression of Aβ42 nucleation. Of note, in two AD mouse models using immunoaffinity purification-mass spectrometry, adduct formation between dDDC and brain Aβ was observed in a similar manner as reported in vitro. The present findings unraveled the lysine-targeting inhibitory mechanism of metastable dietary ingredients regarding Aβ oligomerization.
Introduction
Alzheimer's disease (AD) is the most prevalent neurodegenerative disease. Senile plaques in the brain parenchyma of patients with AD are mainly composed of 42-mer amyloid β protein (Aβ42), whereas vascular deposits largely consist of Aβ40. Both forms are secreted from the Aβ precursor protein (APP) to the extracellular space. They aggregate by forming a β-sheet structure to induce neurotoxicity.1,2 Although Aβ aggregates through oligomerization to generate fibrils as a final form, the oligomers of both isoforms are more neurotoxic than the corresponding fibrils, and Aβ42 is more neurotoxic than Aβ40, identifying Aβ42 oligomers as the primary targets for drug development. “Oligomer” is a broadly defined term used in this work for soluble assemblies in aqueous solution ranging from dimers to large assemblies (∼150-mer); on the contrary, insoluble aggregates are termed fibrils.3,4A nucleation-dependent polymerization model has been used to explain Aβ aggregation in vitro, namely two-phase process of nucleation and subsequent elongation, followed by a plateau phase in which the amount of aggregates reaches an equilibrium5–7 (Fig. S1, ESI†). Soluble oligomers as metastable intermediates of Aβ aggregates that correspond to products of the nucleation phase more strongly contribute to AD etiology than insoluble fibrils as the end product of aggregation.3,4,8,9 Therefore, the development of oligomerization and/or nucleation modulators is a promising approach for delaying the development of toxic oligomers, redirecting the self-assembly process toward dissociation back to less toxic monomers, or in some cases accelerating the process into less toxic fibrils10,11 for AD treatment.
Several showcase and extensive reviews have discussed naturally occurring products exerting anti-aggregation effects through any of the aforementioned mechanisms.12–14 Our group has also reported many natural products from crude drug and functional food components involved in the inhibition of Aβ42 aggregation,15 and the underlying mechanism offers three structural features: [1] the catechol structure in flavonoids that can form Michael adducts or Schiff bases upon autoxidation with side chains of Lys16 and Lys28;16 [2] the conjugated flat structure originating from α,β-unsaturated carbonyl groups and aromatic rings that can interact with the side chains of Phe19 and Phe20;17,18 and [3] a carboxy group structure in triterpenoid or anthraquinoid that can generate a salt bridge with basic amino acid residues of Lys16 and Lys2819 (Fig. S1, ESI†). Pyrone moieties as Michael acceptors expanded from catechol structures can contribute to the suppression of Aβ42 aggregation.20,21 Repositioning warfarin with 2-pyrone to delay the nucleation phase of Aβ42 through Michael addition is a promising approach to identify anti-AD drugs because warfarin is widely used as coagulant drug.11 However, there have been no validation studies regarding whether small ligands covalently bind to brain Aβ in the pathological condition of AD.
Attention must be paid to natural products in water or body fluid condition because such compounds can be readily decomposed. One example with potential use for AD treatment is curcuminoid from Curcuma longa L., which has been suggested to inhibit Aβ aggregation both in vitro22 and in vivo23 and to cross the blood–brain barrier (BBB) in mice,23 fulfilling clinical studies. However, most of curcumin consumed by animals is subject to chemical degradation processes such as solvolysis,24,25 autoxidation,26 and photodegradation,25 and thus, the likely false activity of curcumin has been controversial given evidence that curcumin is a metastable, reactive, and nonbioavailable compound.27
Akaike and colleagues reported that 2′,3′-dihydroxy-4′,6′-dimethoxychalcone (DDC, Fig. 1A), a component of green perilla (Perilla frutescens var. crispa f. viridis), induced cellular resistance to oxidative stress through activating the nuclear factor erythroid 2-related factor 2 (Nrf2)–antioxidant response element (ARE) pathway,28 and subsequently, DDC prevented neurotoxicity in primary cortical neurons deduced from Aβ42 aggregates.29 However, activation of the Nrf2–ARE pathway by DDC did not lead to neuroprotective effects, and DDC pre-incubation was required for neuroprotection,29 prompting us to consider the direct contact of Aβ42 oligomers with DDC or its decomposition forms as the underlying mechanism rather than Nrf2–ARE pathway activation. This study revealed the site-specific inhibitory mechanism of DDC against Aβ42 aggregation through which DDC is transformed into its decomposition forms (dDDC [1–3], Fig. 2) through nucleophilic aromatic substitution with water molecules using a combination of LC-mass spectrometry (MS)-based metabolomics, NMR perturbation experiments, and immunoaffinity purification-MS in AD mouse models. We also discussed the significance of considering biodegradability for AD prevention and treatment, and structural insight into designing specific inhibitors for Aβ oligomerization was obtained.
 | ||
| Fig. 1 Characterization of DDC for its anti-Aβ42 aggregation properties. (A) Structure of DDC. (B) Th-T aggregation assay. Aβ42 (25 μM) and DDC (0.1–100 μM) were incubated in PBS at 37 °C. Data are expressed as the mean ± s.d. (n = 8). (C) Sedimentation assay. Aβ42 (25 μM) and DDC (100 μM) were incubated in PBS at 37 °C. Data are presented as means ± s.d. (n = 2). (D) TEM analysis of aggregates generated from Aβ42 (25 μM) in the presence and absence of DDC (100 μM) after incubation for 48 h at 37 °C. Scale bar = 200 nm. (E) Secondary structure analysis by CD measurement. Aβ42 (25 μM) treated with HFIP and DDC (100 μM) were incubated in PBS at 37 °C. (F) The effect of TCEP, a reductant, on Aβ42 aggregation. Aβ42 (25 μM) and DDC (100 μM) was incubated in the presence and absence of TCEP (100 μM) at 37 °C. Green shadows indicate the nucleation phase, where the relative aggregation of Aβ42 is 50%. | ||
 | ||
| Fig. 2 LC-MS profile of the decomposition products of DDC and the proposal of their structures. (A) The UPLC chromatogram of DDC after its incubation in the buffer together with the measured mass of the generated compounds from DDC [e.g. the closed form (cDDC), the decomposition products (dDDC, 1–3), the oxidized form, the reduced form of olefin, and the methylated form]. After DDC (50 μM) was incubated in 25 mM ammonium acetate (pH 7.4) at 37 °C for 4 h, an aliquot was subjected to LC/Q Tof-MS. (B) Proposal of the structures of dDDC. DDC was supposed to be converted into dDDC through nucleophilic aromatic substitution with water molecule to the unstable intermediates [DDCa or DDCb (−14 Da)]. Further processing of MS/MS spectrometry by FlavonoidSearch suggested three possible flavonoids (1–3), whose molecular weight was identical to the chalcone form (DDCa or DDCb), by intramolecular cyclization. These candidates can form the corresponding Schiff base with Aβ42-Lys16,28. | ||
Results
Inhibition of the aggregation and β-sheet transformation of Aβ42 by DDC through its autoxidation
First, we synthesized DDC as previously described.30 To explore the effect of DDC on Aβ42 aggregation, a fluorescence assay using thioflavin-T (Th-T), one of the most common probes for monitoring the aggregation of generic amyloidogenic proteins,31 was conducted. As presented in Fig. 1B, DDC inhibited the aggregation of Aβ42 (25 μM) in a concentration-dependent manner (0.1–100 μM) during incubation for 24 h, and the IC50 was calculated as 10.6 ± 1.3 μM via nonlinear regression (Fig. S2, ESI†). Comparing to the previously reported IC50s of flavonoids,16 DDC exhibited similarly strong anti-aggregative ability as dietary polyphenols (e.g., morin, 30.3 μM; taxifolin, 33.0 μM). There is inherent bias of the Th-T assay when evaluating the anti-amyloidogenic properties of compounds because of competitive binding with Th-T, as exemplified by (−)-epigallocatechin-3-gallate,32 curcumin,33 and resveratrol.33 To validate the potential anti-aggregative activity of DDC, a sedimentation assay using HPLC after centrifugation was performed. After incubation for approximately 1.5 h at 37 °C, Aβ42 started to nucleate, eventually reaching a plateau after 4 h (Fig. 1C). Conversely, the aggregation of Aβ42 was delayed after 2 h of incubation in the presence of DDC, and Aβ42 treated with DDC did not completely aggregate after 24 h. This result was in good agreement with that of the Th-T assay (Fig. 1B). Using transmission electron microscopy (TEM) to further investigate the morphology of the aggregate, the bundles of amyloid fibrils generated from Aβ42 in the presence of DDC were sparse, and the short fibrils are observed (Fig. 1D), and their width was shortened (Fig. S3A, ESI†).We also examined the effect of DDC on the secondary structure of Aβ42 using circular dichroism (CD) spectroscopy. As presented in Fig. 1E, the intensities of the positive peak at 195 nm and negative peak at approximately 210 nm increased when Aβ42 was incubated alone at 37 °C, indicating transformation into a β-sheet. By contrast, in the presence of DDC, the signal intensities of Aβ42 were weaker than those of Aβ42 alone even after 24 h of incubation. These results suggest that the preventive effects of DDC on Aβ42 nucleation are related to disturbance of the β-sheet transformation of Aβ42.
Autoxidation of catechol moieties to o-quinone is essential for preventing Aβ aggregation.16 As presented in Fig. 1F, the inhibitory effects of DDC against Aβ42 aggregation in the presence of tris(2-carboxyethyl)phosphine (TCEP) as a reducing agent were diminished, especially in the nucleation phase (approximately a 2 h incubation), implying a role of DDC autoxidation in the inhibition of Aβ42 oligomerization.
Structural determination of DDC decomposition products (1–3)
It is well known that chalcones can undergo degradation, demethylation, autoxidation, or isomerization to flavonoids.34 In an attempt to determine the structure of dDDC, DDC pre-incubated in ammonium acetate buffer for 4 h at 37 °C was subjected to LC-MS/MS. Total ion chromatography (TIC) revealed several peaks in addition to that of intact DDC (Fig. 2A). To survey the metabolomics of the chalcone, the obtained information on MS was processed and annotated by FlavonoidSearch,35 which is a database of probable mass fragments for >6000 flavonoids (FsDatabase) for automatic annotation adhering to the MS/MS-aglycone rule using the mass spectra of decomposition peaks as queries with the assistance of CFM-ID as a general identification tool for metabolites.36 These analyses deduced the intact form of DDC (m/z 301.1074, [M + H+] calcd for C17H11O5 300.0908) in addition to the closed form of DDC (cDDC, Fig. 2B) as a flavanone (m/z 301.1074, [M + H+] calcd for C17H11O5 300.0908), dDDC (−14 Da; m/z 287.0909, [M + H+] calcd for C16H15O5 287.0919), and the methylated form of DDC (m/z 315.1238, [M + H+] calcd for C18H19O5 315.3370; Fig. 2A). These data suggested three possible decomposed flavonoids (−14 Da; 1–3 as dDDC) of DDC (chalcone), which might be generated contingent upon nucleophilic aromatic substitution with water molecules and subsequent cyclization of the corresponding chalcones (DDCa and DDCb) to flavonoids (Fig. 2B).Evaluation of DDC and 1–3 for their inhibitory activities on Aβ42 fibrillization and oligomerization
Compounds 1 and 3 were newly identified compounds, whereas 2 was previously isolated from cheilanthoid fern (Notholaena neglecta)37 and green perilla leaves,38 respectively, with low yields. To characterize the contribution of these decomposition products (1–3) to the inhibitory activity of DDC on Aβ42 oligomerization, 1–3 were synthesized using conventional approaches (Scheme S1, ESI†), and were subjected to the Th-T aggregation test. The aggregation of Aβ42 (10 μM) mixed with each compound (50 μM) was monitored in the presence of Th-T reagent (20 μM). To characterize the effect of each compound on Aβ42 nucleation, Aβ42 treated with 1,1,1,3,3,3-hexafluoro-2-propanol (HFIP) was used. In the competitive assay to assess the inherent fluorescence of 1–3, we did not observe significant interference with Th-T fluorescence upon the binding of Aβ42 aggregates to 1–3 as well as DDC (Fig. S4A, ESI†). Furthermore, the inherent potent fluorescence of any compound itself over Th-T fluorescence was not observed (Fig. S4B, ESI†).As presented in Fig. 3A, Aβ42 required approximately 3 h for nucleation and reached a plateau after approximately 8 h. Given the slightly faster velocity of nucleation in Fig. 1B, C and F than in Fig. 3A, the inconsistency in the aggregation curve of Aβ42 might originate from differences in incubation conditions such as temperature, HFIP treatment, and co-incubation with Th-T. Both 1 and 3 slightly delayed the nucleation of Aβ42 and suppressed aggregation in a similar manner as DDC. The inhibitory effects of 3 were significantly stronger than that of 2 after 3 h of incubation. Treatment of Aβ42 with 1–3 produced the decreased amount of amyloid fibrils compared to no treatment (Fig. 3B). The width of Aβ42 fibrils was shortened by incubating with each flavonoid in spite of no significant differences among the width of fibrils treated with each flavonoid (Fig. S3B, ESI†). By contrast, the inhibitory effect of cDDC, which is one of the major products of DDC during incubation (Fig. 2A) was prepared by cyclization of 1 by acetic acid treatment, on Aβ42 aggregation was weaker than that of DDC (Fig. S5A, ESI†), and fibril formation was partially observed in the presence of cDDC (Fig. S5B, ESI†). The fibril width did not significantly alter by adding cDDC (Fig. S3C, ESI†). The IC50 of 1–3 were deduced to be 33.4 ± ![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 1.4, 55.3 ± 2.0, and 14.0 ± 1.2 μM, respectively (Fig. S2, ESI†).
1.4, 55.3 ± 2.0, and 14.0 ± 1.2 μM, respectively (Fig. S2, ESI†).
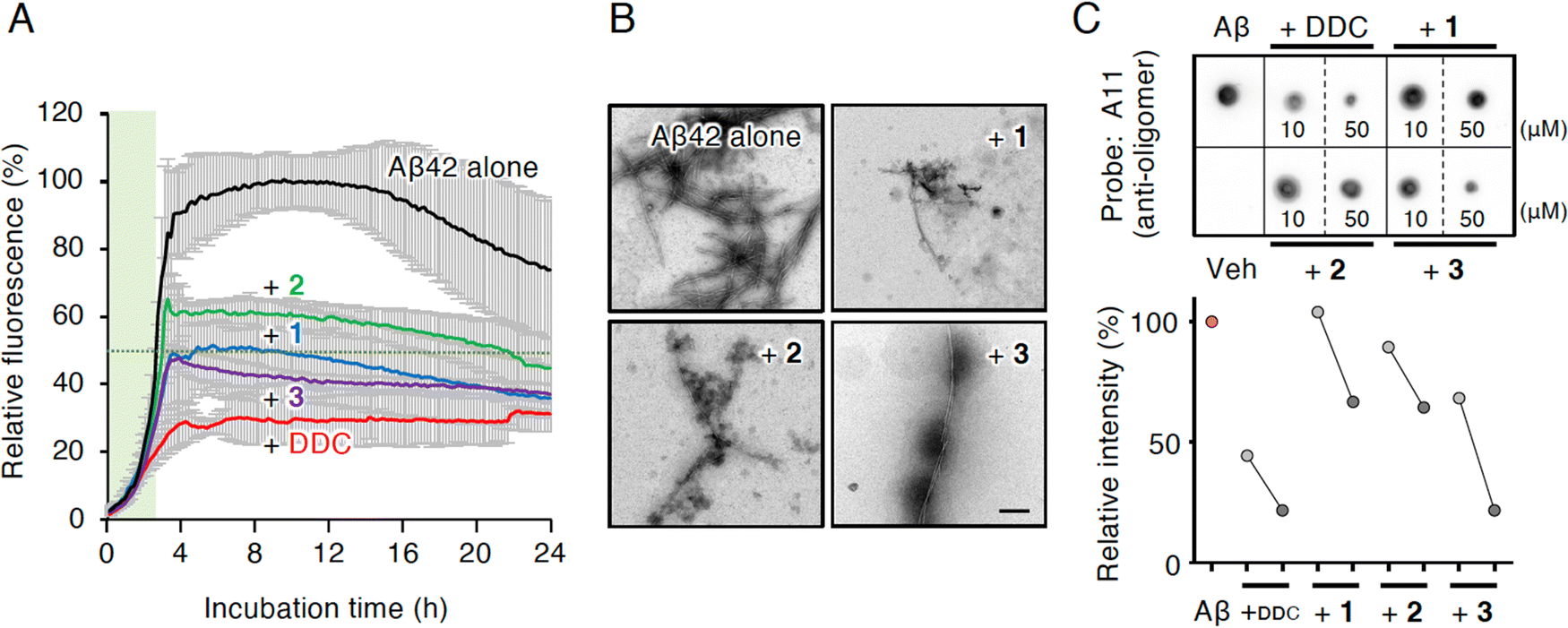 | ||
| Fig. 3 Inhibitory effects of DDC and 1–3 on the fibrillization and oligomerization of Aβ42. (A) Th-T test of DDC and 1–3 against Aβ42 aggregation. Time response curves of aggregation of Aβ42 (10 μM) treated with HFIP during incubation for 24 h at 25 °C in the presence of each compound (50 μM) are indicated. Data are presented as the mean ± s.d. (n = 3). Green shadows indicate the nucleation phase, where the relative aggregation of Aβ42 is 50%. (B) TEM analysis of Aβ42 aggregates incubated with DDC and 1–3 after Th-T test. Scale bar = 200 nm. (C) Dot blotting of Aβ42 using an oligomer-specific antibody (A11). After Aβ42 (10 μM) was incubated with DDC and 1–3 (10 or 50 μM) for 1 h at 37 °C, an aliquot (1 μL, 45 ng) was used for the analysis. Relative intensity to Aβ42 alone of dot blots in the upper panel for 10 μM of each compound for light gray circle and 50 μM of each compound for dark gray circle are indicated in the lower panel. Veh, vehicle. | ||
In an attempt to clarify the ability of DDC and 1–3 to delay the nucleation of Aβ42, we performed dot blotting using an oligomer-specific antibody (A11),39 the specificity of which for Aβ42 oligomer versus the monomer and fibril was previously confirmed by our group.11 Although A11-immunoreactive oligomers were readily detected after the incubation of Aβ42 alone (10 μM) for 1 h, the signals for oligomers were decreased in the presence of DDC and 1–3 (Fig. 3C). Indeed, the signals in the presence of 1–3 were generally decreased in a concentration-dependent manner (10 and 50 μM), and in 3 displayed stronger inhibitor effects than 1 and 2. These findings indicate that the delay of the nucleation phase might be related to mitigation of Aβ42 oligomerization.
NMR analyses of the interacting residues of Aβ42 with DDC and 1–3
To investigate the mechanism by which DDC and 1–3 associate with full-length Aβ42, 1H–15N heteronuclear multiple quantum coherence (HMQC) NMR analysis was performed by focusing on the chemical shift perturbations of uniformly 15N-labeled Aβ42, which generally remained in the monomeric state following pretreatment with HFIP. To suppress the aggregation velocity of Aβ42 during the measurement, we adopted band-selective optimized-flip-angle short-transient (SOFAST)40 HMQC at 4 °C, which gave 2D spectra in approximately 30 min (Fig. 4). The assignment of the chemical shifts of Aβ42 alone was in good agreement with previous reports41–43 including our studies,17–19 excluding the overlapping cross-peaks (Ala2/Ala30 and Val24/Ile31) and missing cross-peaks (His6) as observed previously44 (see also the full spectra in Fig. S6, ESI†).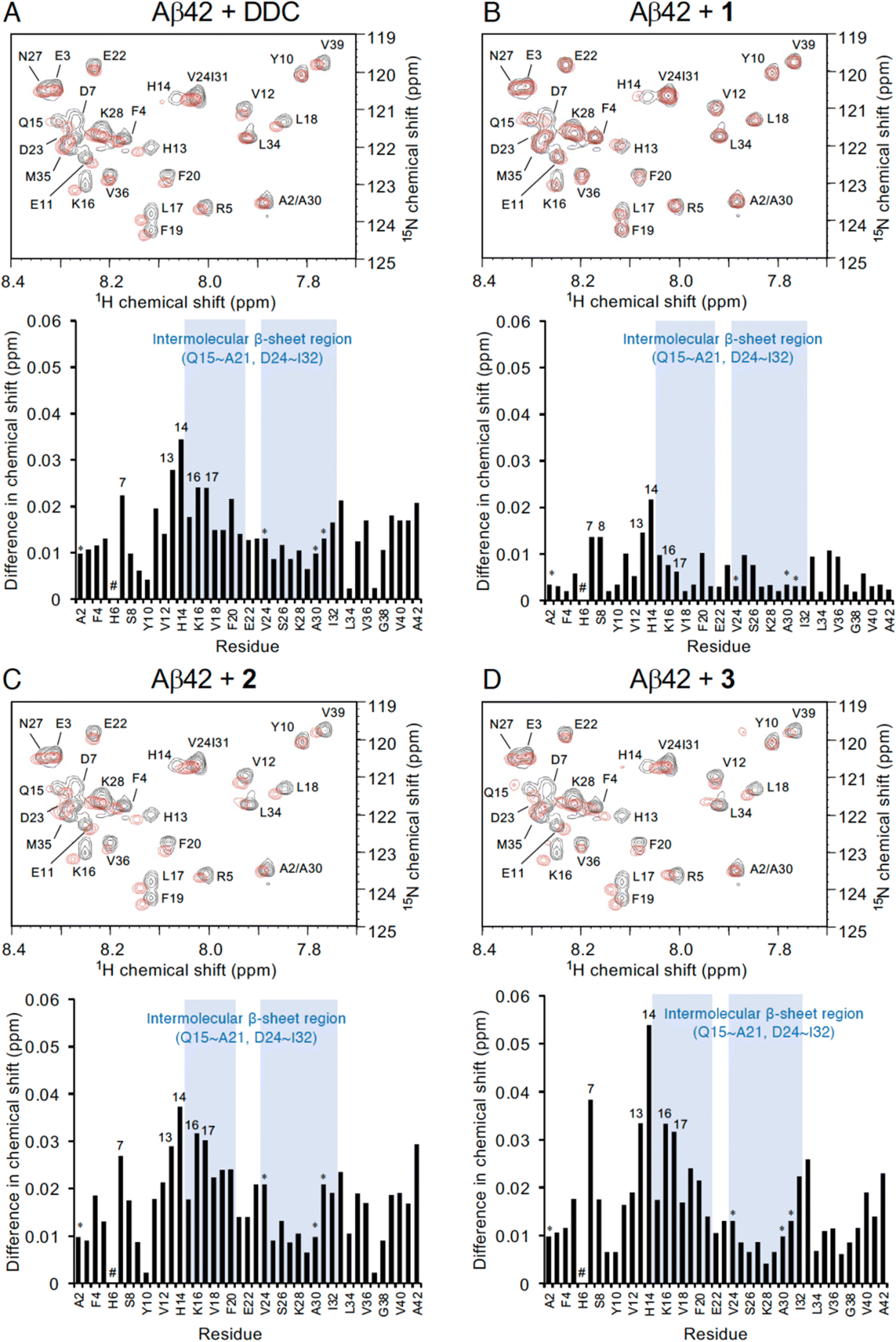 | ||
| Fig. 4 NMR analyses of the interaction of Aβ42 with DDC and 1–3. Expanded 1H–15N SOFAST-HMQC spectra of Aβ42 in the absence or presence of (A) DDC, (B) 1, (C) 2, and (D) 3. Aβ42 uniformly labeled with 15N (25 μM) after HFIP treatment was either treated with each compound (250 μM) or left untreated in PB (pH 7.4), and measurements were conducted for approximately 30 min at 4 °C. Upper, the superimposed HMQC spectra. Black cross peaks, Aβ42 alone; red cross peaks, Aβ42 treated with each compound. Lower, 1H–15N chemical shift differences in HMQC spectra between Aβ42 alone and Aβ42 treated with each compound calculated by the Pythagorean theorem. * The cross peaks of residues (Ala2/Ala30 and Val24/Ile31) overlapped. # The cross peaks of His6 and Asp7 not observed. Full spectra of each measurement are shown in Fig. S6 (ESI†). | ||
In light of adding DDC and 1–3 to Aβ42, Fig. 4 presents the superimposed HMQC spectra of Aβ42 alone (25 μM) and Aβ42 treated with DDC and 1–3 (250 μM). The chemical shifts of Asp7, His13, His14, Lys16, and Leu17 were largely perturbed when Aβ42 was treated with DDC or 1–3 (Fig. 4), and in particular, the extent of perturbation by 3 was comparable to that of DDC (Fig. 4A and D). These perturbed regions were included in the intermolecular β-sheet in the S-shaped fold of ex vivo Aβ42 aggregates from AD brains, as revealed by recent solid-state NMR45–47 and cryo-EM48 studies, which is also important for Aβ42 oligomerization.49 His13 and His14 could induce Aβ42 neurotoxicity by chelating copper or zinc ions to produce a phenoxyl radical at Tyr10.50,51
Formation of a Schiff base by DDC with Lys16 and Lys28 in the β-sheet region of Aβ42 via autoxidation
Although 1–3 and DDC interacted with Aβ42 (Fig. 4), DDC can easily undergo nucleophilic aromatic substitution to 1–3 in the buffer, followed by intramolecular cyclization. To further determine the molecular interaction between Aβ42 and dDDC, we subjected a mixture of Aβ42 and DDC in ammonium acetate buffer after 10 min of incubation at 37 °C to liquid chromatography/quadrupole time-of-flight mass spectrometry (LC/Q-Tof-MS). As presented in Fig. 5A, possible peaks of Aβ42 adducts with dDDC (−14 Da, tR = 7.22 min) were detected along with Aβ42 (tR = 7.35 min) in TIC. The mass envelope at charge distribution (tR = 7.22 min) corresponded to the Schiff base, but not the Michael adduct, of Aβ42-Lys16/Lys28 with DDC (Fig. 5B, deconvoluted mass: 4779.68, calcd: 4780.39). Another peak (tR = 7.35 min) originated from the unreacted Aβ42 (Fig. 5A, deconvoluted mass: 4514.95, calcd: 4514.14). It is of interest to note that the mixture of Aβ42 and DDC underwent spontaneous autoxidation to generate the Aβ42 adduct with dDDC, which could be required to prevent Aβ42 aggregation. Indeed, no adduct peaks for Aβ42 with intact DDC were observed, as exemplified by apomorphine, which was easily autoxidized to form the Michael adduct of Aβ42-Lys16/Lys28 with the oxidized apomorphine.18 The flavonoid forms (1–3) of DDC proposed in Fig. 2B are suggested to form the corresponding Schiff base with Aβ42-Lys16/Lys28. | ||
| Fig. 5 Formation of Aβ42 adduct at Lys16,28 with DDC in LC-MS/MS analysis. (A) LC/Q Tof-MS analysis of Aβ42 treated with DDC, showing TIC (left) and the deconvoluted mass spectrometry (middle and right) of the adduct of Aβ42 treated with DDC (tR = 7.22 min) and unreacted Aβ42 (tR = 7.35 min) indicated as arrows. After Aβ42 (25 μM) and DDC (50 μM) was incubated in 25 mM ammonium acetate (pH 7.4) at 37 °C for 10 min, an aliquot was subjected to the analysis. (B) LC/Q Tof-MS analysis of the E22P,M35ox-Aβ9-35 as a model of toxic conformer of Aβ42 treated with DDC, showing UV at 220 nm (left) and measured mass (right) of Aβ adduct treated with DDC (tR = 8.88 min). The relative intensities of mass spectra to a reference are indicated. After E22P,M35ox-Aβ9-35 (25 μM) was incubated with DDC (50 μM) in 50% acetonitrile containing 0.01% NH4OH at 37 °C for 10 min, an aliquot was subjected to the analysis. (C) MS/MS profile of E22P,M35ox-Aβ9-35 with DDC in the measurement of (B). Shown are the sequences of (upper) Lys16 or (lower) Lys28 of E22P,M35ox-Aβ9-35 with DDC. The lines between amino acids show the observed fragment ions, corresponding to the +2 charged peaks labeled as ‘b’ (N-terminal fragment) ions and those labeled as ‘y’ (C-terminal fragment) ions. The precursor ion in expanded MS/MS spectra was set at 1071.2 Da, corresponding to adduct of E22P,M35ox-Aβ9-35 with DDC as a +3 ion in (B). | ||
To characterize the adduct of DDC with Aβ42 and specific amino acids involved in its interaction, we employed LC-MS/MS with collision-induced dissociation, an alternative fragment peptide (E22P,M35ox-Aβ9-35) derived from Aβ42 as a toxic conformer surrogate52 because it was difficult to assign all of the observed fragments from full-length Aβ42. As presented in Fig. 5B, possible peaks of the E22P,M35ox-Aβ9-35 adduct resulting from Schiff base formation with dDDC was detected with the mass envelop at charge distribution (deconvoluted M + H+: 3211.54, calcd: 3210.69) together with unreacted E22P,M35ox-Aβ9-35. A large number of fragmented b and y ions as the +2 charged peaks were detected, yielding a high degree of confidence in the sequence identification of E22P,M35ox-Aβ9-35 and its Schiff base with dDDC at Lys16 or Lys28 (Fig. 5C).
Inhibition of Aβ42 oligomerization by DDC via association with the dimer
To study the suppressive mechanism of DDC in the nucleation phase of Aβ42, ion mobility (IM)-MS was performed to characterize the early oligomeric profile of Aβ42 with DDC. Avoiding the disruption of noncovalent interactions among amyloid oligomers by avoiding organic solvents and using nano-electrospray ionization enabled us to observe the nearly native status of amyloid oligomers in the presence of aggregation inhibitors.53,54 After deconvolution based on the observed mass, peaks corresponding to oligomeric orders of Aβ42 were assigned to the series of multivalent ions depending on their drift time (dt). The peaks for the dimer and trimer in addition to the monomer were apparently observed immediately after dissolution with the buffer (Fig. 6A). By contrast, the addition of DDC led to the disappearance of some of these oligomer peaks, and the adduct peaks of the monomer and dimer with dDDC (−14 Da) were found (Fig. 6A). The two-dimensional heat map composed of the domain of m/z and dt revealed that the adduct peaks of Aβ42 with dDDC adjacent to unbound Aβ42 peak were observed after adding DDC (Fig. 6B).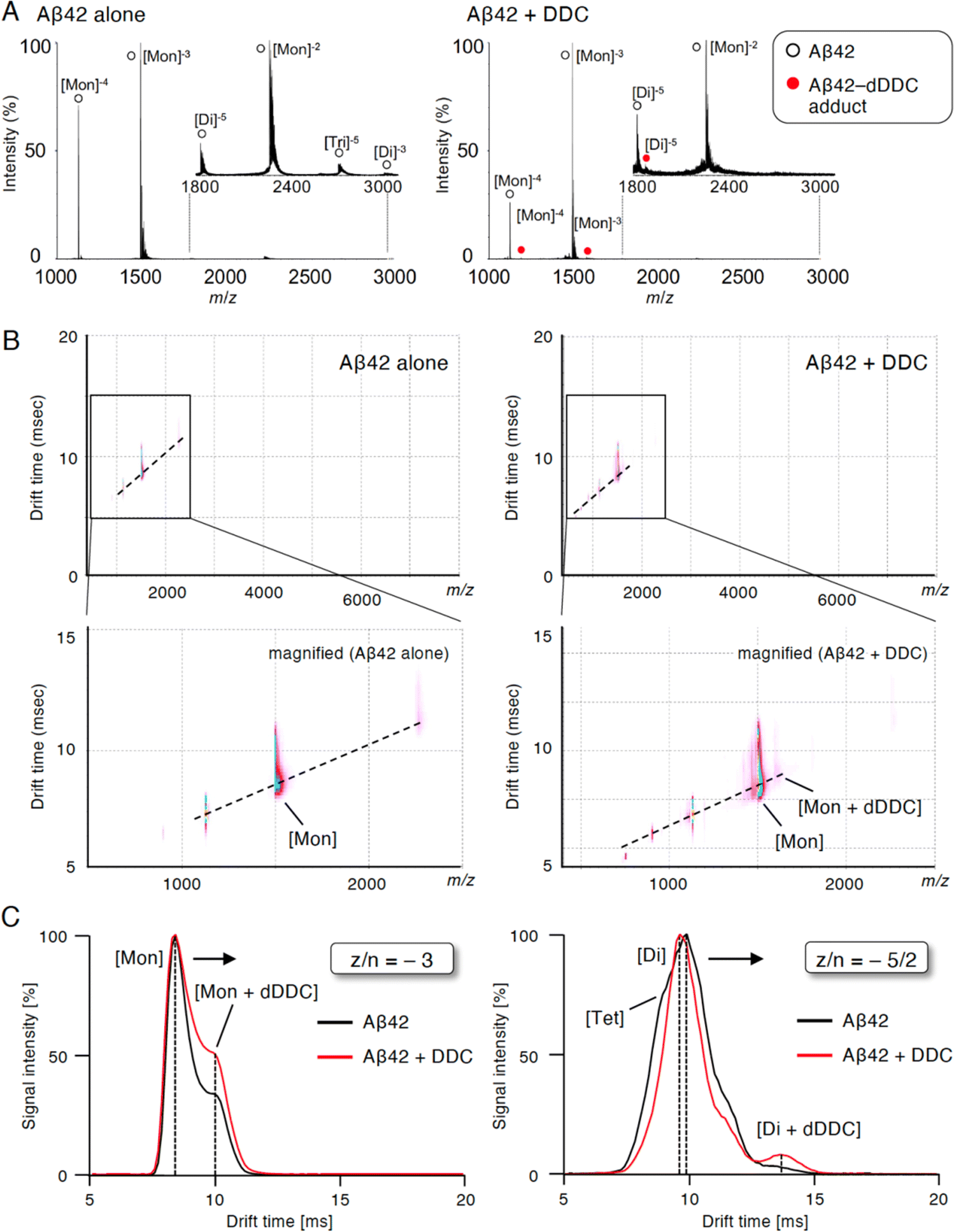 | ||
| Fig. 6 Inhibition of Aβ42 oligomerization by associating with the dimer by DDC. (A) NanoESI-TOF-MS of Aβ42 treated with DDC. Immediately after Aβ42 (40 μM) was mixed with DDC (80 μM) in 25 mM ammonium acetate (pH 7.4), an aliquot was subjected to the analysis. Peaks for Aβ42 and Aβ42–DDC complex are noted with white and red circles, respectively. (B) The two-dimensional heat map composed of the domain of m/z and drift time of IM–MS of Aβ42 treated with DDC. Immediately after Aβ42 (40 μM) was mixed with DDC (80 μM) in 25 mM ammonium acetate (pH 7.4), an aliquot was subjected to the analysis. The signal amplitude with linear scale is color-coded, increasing from purple (low intensity) to blue (high intensity). The area in the rectangle is shown magnified in below panel. (C) Drift time in IM–MS for z/n = −3, and −5/2 peaks of Aβ42 without (black) or with (red) DDC. z/n = charge/oligomer number. The number of units such as monomer (Mon), dimer (Dim), trimer (Tri), and tetramer (Tet) was determined by their drift time. | ||
The arrival time distribution (ATD) of z/n = −3 ions (z/n = charge/oligomer number) displayed one distinct peak representing the monomer (dt = 8.4 ms) with Aβ42 alone, whereas additional peaks representing a more extended conformation of the monomer including the monomer + dDDC adduct (dt = 9.9 ms) appeared when Aβ42 was treated with dDDC (Fig. 6C). Moreover, the ATD of z/n = −5/2 ions revealed one predominant peak as the dimer (dt = 9.5 ms) for Aβ42 alone based on the deconvolution analysis (Fig. 6C). In the presence of DDC, dt of the dimer + dDDC adduct (dt = 13.4 ms) was largely retarded compared to that of the dimer, resulting in the change of the oligomeric conformation to an extended state by DDC and the suppression of further oligomerization (n ≥ 3). Therefore, DDC could generate a Schiff base with Lys16 or Lys28 in the β-sheet region of the Aβ42 monomer or dimer via conversion to 3, resulting in the prevention of higher-order oligomer formation.
Detection of Aβ42 adducts with dDDC in the brains of AD mouse models
To assess whether Aβ can also form adducts with DDC in vivo, we performed immunoprecipitation combined with MS (IP-MS) using aged Tg2576/PS2 mice (9 months old)55 as an animal model of AD that predominantly produces Aβ40 together with Aβ42 in the brain. Soluble Aβ was extracted by homogenizing the brain extracts of AD mouse models treated with DDC in a buffer for 30 min without detergents to avoid hampering the Aβ–ligand interaction. A previous IP-MS study using 6E10 antibody targeting Aβ1–16 revealed that several Aβ cleavage products such as Aβ11–40 and other unidentified fragments in addition to Aβ1–X (X = 33, 34, 37, 38, 40, 42, and 43) in the brains of Tg2576 mice.56 Thus, to characterize the profile of Aβ1–X, in which Asp1 was retained, the antibody (82E1) against the N-terminus of Aβ was used for immunoprecipitation, followed by analysis by matrix-assisted laser desorption/ionization time-of-flight mass spectrometry (MALDI-TOF-MS). We predominantly detected Aβ38, Aβ40, and Aβ42 in the vehicle treatment group, and shorter Aβ cleavage products were less frequently observed (Fig. 7A). It should be noted that the adducts of both Aβ40 and Aβ42 with dDDC in addition to the intact Aβ were detectable (Fig. 7A).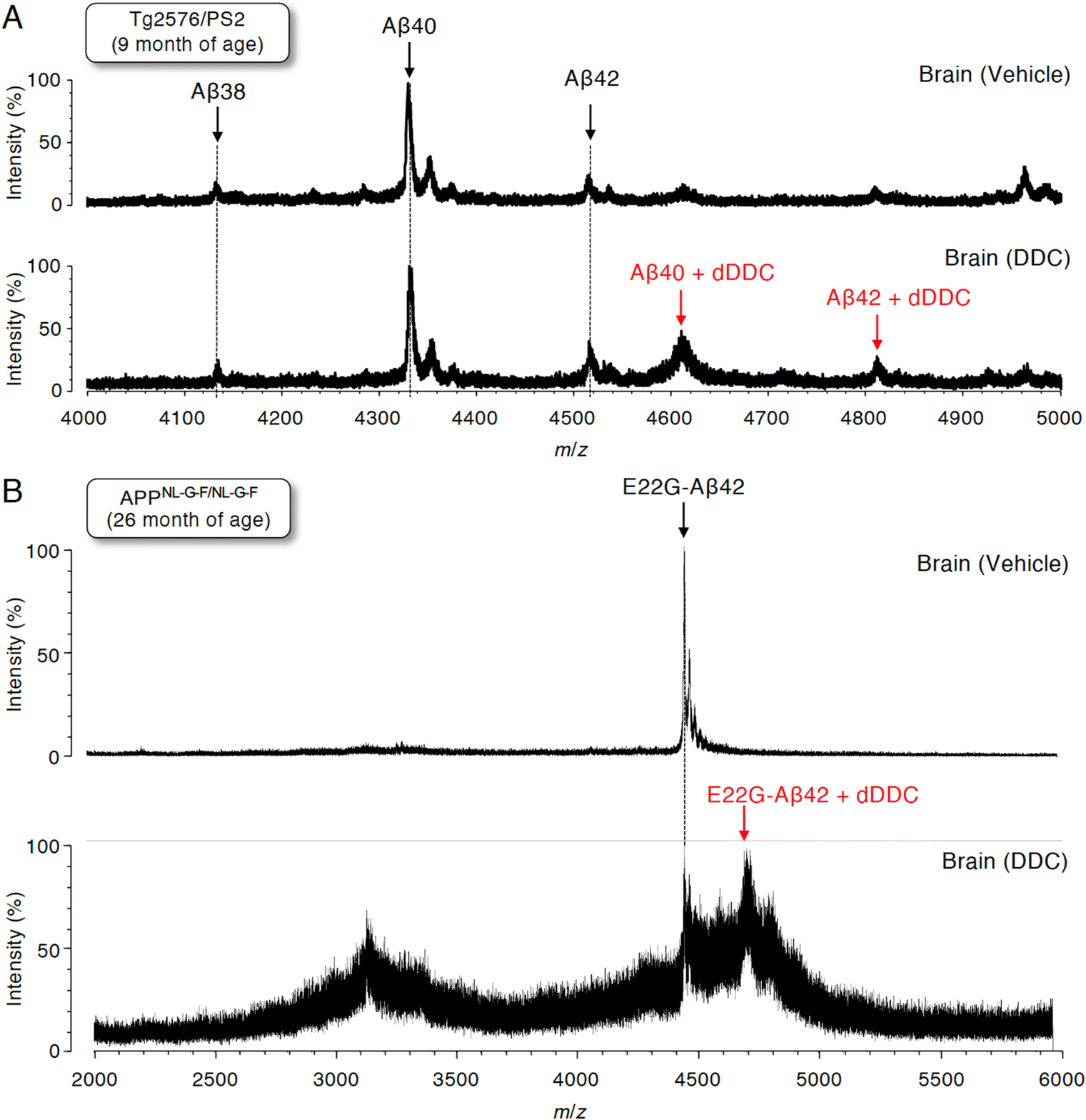 | ||
| Fig. 7 MALDI-TOF-MS profile of brain Aβ immunoprecipitated from two lines of AD mouse models fed with DDC. DDC was orally administered to (A) Tg2576/PS2 mice at the age of 9 months and (B) APPNL-G-F/NL-G-F mice at the age of 26 months. After 30 min of administration, full-length Aβ starting at Asp1 was extracted by brain homogenization using Tris–HCl buffer without the detergent, and the subsequent immunization precipitation using the anti-N-terminus of the Aβ (82E1) antibody. Adducts of Aβ40 and Aβ42 with dDDC in addition to unreacted Aβ38, Aβ40, and Aβ42 were observed in (A), and E22G-Aβ42 adducts with dDDC in addition to unreacted E22G-Aβ42 were observed in (B), respectively. | ||
Given the preferable formation of protofibrils (PFs), which are toxic oligomers, by E22G-Aβ42,57 APPNL-G-F/NL-G-F knock-in mice58 preferably producing E22G-Aβ42 (arctic mutation) represent an oligomer-generating aged model. We also used aged APPNL-G-F/NL-G-F (26 months old) for IP-MS experiments. Similarly, using 82E1 antibody for immunoprecipitation, we mainly observed E22G-Aβ42 in the vehicle treatment group (Fig. 7B). Treatment with DDC induced the adduct formation of E22G-Aβ42 with dDDC together with Aβ (Fig. 7B). Ion suppression of Aβ signals by adduct formation in the DDC-treated old mice (26 months old: Fig. 7B) might be due to its lower amount of soluble brain Aβ than that of the DDC-treated young mice (9 months old: Fig. 7A).
Discussion
As exemplified by the ongoing curcumin debate,59 the metastability of highly reactive food compounds and the compound structure that is responsible for biological activity are important interests for drug development using natural products. Despite difficulty in the comprehensive identification or annotation of flavonoids,60,61 flavonoids can be modified under physiological conditions such as those in the intestine or liver, and in particular, it is worth noting that the conversion of chalcones to flavonoids plays a role in the anti-aggregation activity of DDC, and 3 could be one of the active forms of DDC. The current findings provided structural insight into the inhibition of Aβ42 oligomerization by DDC through its converting to dDDC via nucleophilic aromatic substitution with water molecules, and Lys residues in Aβ42 could be specifically targeted by forming a Schiff base with the decomposed forms through autoxidation of dDDC. Overall, comprehensive MS studies coupled with synthetic approaches deciphered that 3 is one of the most responsible decomposed flavonoids among dDDC moieties for the inhibitory activity of DDC. In other words, the trapping of 3 by Aβ42 might maintain its stability. Although 1–3 have never been isolated from green perilla, 3 might exhibit anti-Aβ aggregation activity even at low levels. Alternatively, regardless of the binding activity of DDC with dimers, Aβ42 aggregation partly occurred as shown in Th-T assay, implying that either DDC adducts with dimers could not be enough for complete inhibition of Aβ42 aggregation process or these adducts might be not long-term stable.To our knowledge, we demonstrated for the first time by IP-MS using Tg2576/PS2 mice orally treated with DDC that dDDC generated covalent adducts with both Aβ40 and Aβ42, the latter of which is a minor species of brain Aβ. Further evaluation using APPNL-G-F/NL-G-F mice, which preferentially exhibit PF formation, implied the preferable recognition of DDC by Aβ oligomers. The levels of PFs were reported to correlate with the cognitive impairment of Arctic AD transgenic mice.62 Rats have been used instead of mice to analyze the brain accumulation of oral compounds because of their bigger brain size.63,64 In our study, aging (9 and 26 months) in transgenic mice might have affected the BBB permeability, resulting in the successful detection of Aβ adducts with an orally consumed compound. Indeed, BBB degradation is considered an early biomarker of human cognitive dysfunction.65 Although the characterization of soluble and insoluble Aβ in the transgenic mouse brain as an AD model has been investigated using MS by some groups,66,67 this study successfully identified the Schiff base of an anti-aggregative food component with full-length Aβ probed using an antibody targeting the N-terminus of Aβ in a similar manner as in vitro studies. These rationales of the association of metastable functional food components with Aβ oligomer in vitro and in vivo could support the design and development of anti-AD drugs. However, the N-terminus of Aβ extracted from plasma and small intestine was not deleted, or most of the immunoprecipitated Aβ was degraded (data not shown). Further in vivo experiments are needed to elucidate the pharmacokinetics of Aβ adducts in the digestive system, such as the use of radioisotope-labeled Aβ.
In most cases, the covalent bond formation between Aβ with a small molecule ligand was the formation of a Michael adduct.68 Conversely, this study uniquely observed Schiff base formation between Aβ42 and dDDC even though only one study on the Schiff base of α-synuclein with baicalein has been reported.69 Schiff base formation between Aβ40 fibrils and epigallocatechin-3-gallate was also suggested, but direct observation via MS was not indicated.70 The partial contribution of 2, a non-catechol-type dDDC, to anti-aggregation, supports for Schiff base formation in the inhibitory mechanism of Aβ42 aggregation. Comparisons of the IC50 value each compound in the Th-T test suggested that the anti-aggregation activities of 1 and 3 as catechol-type flavonoids exceeded that of 2 as a non-catechol-type flavonoid, implying the significance of the catechol structure to inhibition. In particular, the regiochemistry of 3 regarding the methoxy and hydroxyl groups on the A-ring was closer to that of DDC. Furthermore, there are reports on the inhibition of Aβ aggregation by 2′,3′-catechol-type flavonoids (e.g., negletein71) such as 3, but not 4′,5′-catechol-type flavonoids (e.g., norwogonin, isoscutellarein) such as 1.
The suppression of the A11-reactive oligomerization of Aβ42 by 3 was associated with its apparent delay of nucleation in a similar manner as DDC. These findings imply the preferable inhibition of toxic oligomers by 3 and supported the hypothesis of the oligomeropathy of Aβ in the pathology of AD.3,4,8,9 Considering our nine-group classification method of natural products based on their ability to modulate the nucleation and/or elongation phases,11 DDC or 3 was categorized into Group I, in which both the nucleation and elongation phases were hampered. These findings did not contradict our rationale that these compounds,15 as exemplified by (R)-apomorphine,18 as all compounds were antitoxic and they contained catechol and flatness, were promising scaffolds for anti-AD drugs. There is a possibility that DDC itself can be degraded into dDDC during the comparison assays with each dDDC, and thus dDDC mixtures may contribute to inhibitory activity of DDC in these assays. Alternatively, the concentration of DDC used in each assay might affect the amount of intact DDC (e.g. 250 μM in SOFAST-HMQC measurement; 50 μM in dot blotting and Th-T assay).
LC-MS-based metabolomics is a powerful tool for the exhaustive analysis of metastable natural products such of DDC with predictable reliability and efficacy.72,73 NMR-based metabolomics can reinforce the structure determination of citrus-derived compounds from their chemical shifts and coupling parameters.74 Computational analysis based on a chemical database such as FlavonoidSearch as performed in this study also facilitated the identification of active compounds with anti-AD properties. Our group recently developed an activity-differential search for Aβ42 aggregation inhibitors from crude drugs using LC-MS combined with principal component analysis by drawing correlations between the extracts of different parts of the same plant and their biological activity.75 These accumulated studies on metastable bioactive natural products have proven useful for drug development.
Recently, preclinical studies by Saito et al. on (+)-taxifolin revealed the pleiotropic neuroprotective effects of taxifolin in cerebral amyloid angiopathy by restoring impaired cerebral blood flow76 and suppressing triggering receptor expressed on myeloid cells 2-associated inflammation,77 apart from its direct association with Aβ42 (Michael adduct formation at Lys16/Lys28).16 Their group is currently preparing a clinical trial of (+)-taxifolin in patients with cerebral amyloid angiopathy.78 Their pharmacokinetic studies on (+)-taxifolin in mice, given by solution orally via gavage with a dose (300 mg kg−1) that was preventive for Aβ accumulation, showed the maximum concentration (Cmax) of blood levels of (+)-taxifolin was 19 μM.76 Although this value does not seem far from the effective concentration in vitro, the optimal dose and usage of (+)-taxifolin in clinical studies for AD should be assessed for safety and tolerance. Indeed no side effects were found in the previous clinical studies on treatment with taxifolin 40–120 mg per day for 2 weeks ∼ 3 months.79 The similar concerns might be applicable to DDC or its decomposed flavonoids. Because DDC also site-specifically targets Lys16/Lys28 in Aβ, (+)-taxifolin and DDC might share a common mechanism. Considering AD as a potential mixed dementia,80 DDC might be promising for preventing aggregation in disease-causing amyloidosis. These findings highlight the need for consideration on off-target effects and dosage regulation.
Experimental procedures
Thioflavin-T (Th-T) fluorescence assay
The aggregative ability of Aβ42 was evaluated by the thioflavin-T (Th-T; Sigma-Aldrich, St. Louis, MO, USA) fluorescence assay using synthesized Aβ42.81,82 In the measurement at several time points as described elsewhere,52 Aβ42 was dissolved in 0.1% NH4OH at 250 μM, followed by a 10-fold dilution with phosphate buffered saline (PBS: 50 mM sodium phosphate and 100 mM NaCl, pH 7.4) to a final concentration of 25 μM. After incubating of Aβ42 solution (25 μM) without or with each sample using 100× concentrated stock at 37 °C for the desired duration, 2.5 μL of the reaction solution was frozen in a 96-well black plate (Thermo-Fisher Scientific, Waltham, MA, USA) and stored at −80 °C. DDC was dissolved in EtOH at 10 mM, and then assayed at 0.1–100 μM. The resulting fluorescence values after a 24 h incubation were plotted against concentration using GraphPad Prism 9.3.1 (GraphPad Software, San Diego, CA) to deduce IC50 value from nonlinear regression. For investigation of the relevance of autoxidation of DDC to its anti-aggregative ability against Aβ42, TCEP was dissolved in distilled water at 10 mM, and used at 100 μM in the assay. For measuring fluorescence, the sample aliquot was added to 250 μL of 5.0 μM Th-T (Sigma) in 5.0 mM Gly-NaOH (pH 8.5) using 1 mM Th-T solution in distilled water as a stock solution, followed by the measurement at 430 nm excitation and 485 nm emission using a Fluoroskan Ascent microplate reader (Thermo-Fisher Scientific). Th-T relative fluorescence was expressed after subtraction of vehicle control without Aβ42.In the continuous measurement for 24 h described elsewhere,19 Aβ42 pre-treated with HFIP in order to remove the unavoidable aggregation seeds during preparation of Aβ42 was used for assay. Aβ42 was mixed in PBS without or with each compound (DDC, cDDC, and 1–3) plus Th-T solution, and the final concentration of Aβ42 (100 μM in 0.1% NH4OH as a stock) and Th-T (1 mM in distilled water as a stock) was adjusted to be 10 and 20 μM in PBS, respectively. The final concentration of each compound was set to be 50 μM from 10 mM stock in EtOH. The solution was aliquoted into a 96-well black plate (Greiner Bio-One, Monroe, NC, USA) before measuring the fluorescence at 430 nm excitation and 485 nm emission (100 μL per well for 96-well) at 25 °C with agitation at 10 min intervals using a microplate reader (Fluoroskan Ascent; Thermo Scientific). Th-T relative fluorescence was expressed after subtraction of vehicle control without Aβ42.
In interference test to examine whether each compound competes to Th-T for binding Aβ aggregates, Th-T fluorescence was measured immediately by adding each compound (50 μM) after incubation of the Aβ42 solution (10 μM) alone at 37 °C for 24 h. In another interference test to examine whether each compound competes to Th-T itself for showing fluorescence, Th-T fluorescence was measured immediately by mixing with each compound (50 μM).
Sedimentation assay
A sedimentation assay using HPLC (Waters model 1525 with a model 2489 UV detector, Waters, Milford, MA, USA) was performed as described previously, with a slight modification.83 Preparation of the Aβ42 solution (25 μM) without or with DDC (100 μM) was the same as that described for the Th-T assay. After incubation at 37 °C for the desired period, each sample was centrifuged at 17900g at 4 °C for 10 min. Then, 25 μL of each supernatant was analyzed by reversed-phase HPLC on a Develosil-packed column (ODS-UG-5, 6.0 mm i.d. × 100 mm), with elution at 1.0 mL min−1 by a 30 min linear gradient of 10–50% CH3CN in 0.1% NH4OH. The absorption peak at 220 nm was integrated into the area proportional to the amount of substance. The value of the area at each period was subtracted from the highest value, and the percentage of the obtained one to the highest one was defined as “insoluble Aβ42”.
Transmission electron microscopy (TEM)
The aggregate of each Aβ solution (25 μM) without or with DDC (100 μM) in PBS after a 48 h incubation in a similar manner to Th-T assay was examined under a TEM (JEM-1400, JEOL, Tokyo, Japan). Alternatively, in the case of 1–3, the aggregate prepared from each Aβ solution after Th-T assay was applied to TEM observation. After each Aβ aggregate was centrifuged (4 °C, 17900g, 10 min), the supernatant was removed from the pellet. The resultant pellet was gently resuspended in water using a vortex for 1 min just before TEM analysis. The sample suspension (15 μL) was applied to a 400-mesh carbon-coated copper grid (thickness: 20–25 nm; Veco, Eerbeek, Netherlands) and incubated for 5 min before being negatively stained twice with 2% uranyl acetate. Stained samples were subjected to TEM. Fibril width was measured using ImageJ (Wayne Rasband, NIH, MD, USA).
CD spectrometry
CD spectra were measured using a 0.1 mm quartz cell on J-805 instrument (JASCO, Tokyo, Japan) as described elsewhere.19 In brief, HFIP-treated Aβ42 solution in 0.1% NH4OH at 250 μM and DDC in EtOH at 10 mM were diluted with PBS to a final concentration of 25 μM and 100 μM, respectively, and were then incubated at 37 °C. After several intervals, an aliquot (200 μL) was loaded into the quartz cell, and the CD spectrum was recorded at 190–250 nm. The spectra of Aβ42 are shown after subtraction of the spectrum of the vehicle alone, and those in the presence of DDC are shown after subtraction of the spectrum for DDC alone.LC-MS/MS of DDC
DDC was dissolved at 10 mM, and then diluted with 25 mM ammonium acetate at 50 μM. The resultant solution was incubated for 4 h at 37 °C. After being centrifuges at 17900g for 10 min at 4 °C, an aliquot (1 μL) of the supernatant was subjected to a liquid chromatography/quadrupole time-of-flight mass spectrometry (LC/Q Tof-MS) on an Acquity UPLC BEH C18 (particle size 1.7 μm, 2.1 mm I.D. × 100 mm, Waters). The mobile phases consisted of solvent A (acetonitrile containing 0.1% formic acid) and solvent B (H2O containing 0.1% formic acid) under the following gradient program: 5% A for 1 min, 5–25% A for 2 min, 25% A for 5 min, 25–55% A for 2 min, and 55% A for 1.5 min at a flow rate of 0.45 mL min−1. LC-MS/MS analysis was performed on an Acquity UPLC H-class coupled with Xevo G2-S (Waters). MS analysis was performed under the following parameters: capillary voltage for ionization, 2.5 kV (positive mode); sampling cone voltage, 30 V; source temperature, 100 °C; desolvation temperature, 300 °C; cone gas flow, 50 L h−1; desolvation gas flow, 800 L h−1. The collision energy was set at 25–40 V.
Flavonoid analysis
The analysis was performed using FlavonoidSearch35 as a free web server for prediction of flavonoid decomposition. The identity of each fragment ion in LC-MS and LC-MS/MS was evaluated using the m/z value with a given mass tolerance. The essential fragments determined by the annotation rule in these web servers were used for prioritization of the search results with an assistance of CFM-ID.36 Candidates with fewer of the essential fragments missing were prioritized higher than those with more of the essential fragments missing at the same similarity score. Flavonoid structures were obtained from one of the largest flavonoid databases (metabolomics.jp) for virtual fragment generation. The candidates were first selected using the precursor m/z from the deposited flavonoids with a given mass tolerance. Mismatches between the structures and flavonoid names were manually curated with reference to the original literature cited at the website.Synthesis of 1–3
Experimental procedures with detailed spectroscopic data are shown in ESI.†Dot blotting
Aβ42 solution (10 μM) was incubated with or without each compound (DDC and 1–3, 10 or 50 μM) at 37 °C for 1 h. 1 μL (45 ng) of each Aβ sample was applied to the nitrocellulose membrane (0.2 μm pore size, Biorad), and the membrane was blocked with Blocking Reagent for Can Get Signal (TOYOBO, Tokyo, Japan). The membrane was then probed with A11 antibody39 at 1 μg mL−1 (Invitrogen, Carlsbad, CA), followed by treatment with the secondary antibody (anti-rabbit IgG or anti-mouse IgG). Development was performed with ECL chemiluminescence (GE Healthcare). The first and second antibodies were diluted with Can Get Signal Solution 1 and 2 (TOYOBO), respectively. Phosphatase buffered saline plus potassium (PBS-K; 1.5 mM KH2PO4, 8.1 mM Na2HPO4, 140 mM NaCl, 2.7 mM KCl, pH 7.4) including 0.5% Tween-20 was used as a washing buffer.1H–15N SOFAST-HMQC NMR measurement
1H–15N SOFAST-HMQC measurement was carried out using a Bruker Avance II 950 MHz spectrometer with TCI CryoProbeTM (Bruker Biospin, Germany) mainly according to our previous report.17 Uniformly 15N-labeled Aβ42 (67.7 μg; rPeptide, Bogart, GA, USA) treated with HFIP was dissolved at 250 μM in 10 mM NaOH, containing 10 mM EDTA. Each compound (DDC and 1–3) was dissolved in EtOH at 10 mM, and 5 μL of the solution was diluted with 165 μL of 5 mM phosphate buffer (PB: 5 mM sodium phosphate, pH 6.98) containing 5% D2O (10 μL). Then, 250 μM of the Aβ42 solution (20 μL) was added, so that the final concentrations of Aβ42 and each compound were 25 μM and 250 μM in 200 μL solution, respectively. Peaks were assigned by referring to previous findings.17,41–43 Each distance in the 1H–15N chemical shifts between the cross peaks of Aβ42 alone and Aβ42 in the presence of each compound was calculated by the Pythagorean theorem. 15N chemical shifts were scaled one-tenth relative to 1H chemical shifts because the measurement range (7.6–8.7 ppm) of observations of 1H was approximately one-tenth of that (107–128 ppm) of 15N.LC-MS analysis of Aβ42 with DDC
The Aβ42 (250 μM in 0.1% NH4OH) and DDC (5 mM in EtOH) was adjusted to be 25 μM and 50 μM in 20 mM ammonium acetate, respectively. The mixture was incubated at 37 °C for 10 min, and centrifuged for 5 min at 17900g at 4 °C. The supernatant (1 μL) was injected into LC/Q Tof-MS on an Acquity UPLC Peptide BEH C18 (particle size 1.7 μm, 2.1 mm I.D. × 150 mm) and eluted by 10 min linear gradient of 10–50% acetonitrile containing 0.1% NH4OH at 0.2 mL min−1. LC-MS analysis was performed on an Acquity UPLC H-class coupled with Xevo G2-S (Waters). The instrument was operated in negative ion mode with a capillary voltage of 2.0 kV, a sample cone voltage of 25 V, a source temperature of 120 °C, a desolvation temperature of 400 °C, a cone gas flow of 50 L h−1, and a desolvation gas flow of 800 L h−1.
LC-MS/MS of E22P,M35ox-Aβ9-35 with DDC
The E22P,M35ox-Aβ9-35 (250 μM in 0.1% NH4OH) and DDC (10 mM in EtOH) was adjusted to be 25 μM and 50 μM in 50% acetonitrile containing 0.1% NH4OH, respectively. The mixture was incubated at 37 °C for 10 min, and centrifuged for 5 min at 17900 g at 4 °C. The supernatant (1 μL) was injected into LC/Q Tof-MS on an Acquity UPLC Peptide BEH C18 (particle size 1.7 μm, 2.1 mm I.D. × 150 mm, Waters) and eluted by a 10 min linear gradient of 10–40% acetonitrile containing 0.1% NH4OH at 0.3 mL min−1. LC-MS/MS analysis was performed on an Acquity UPLC H-class coupled with Xevo G2-S (Waters). The instrument was operated in positive ion mode with a capillary voltage of 3.0 kV, a sample cone voltage of 40 V, a source temperature of 120 °C, a desolvation temperature of 450 °C, a cone gas flow of 50 L h−1, and a desolvation gas flow of 800 L h−1. The collision energy was set at 38 V. The precursor ion was set to be m/z 1071.2 ([M + 3H]3+).
Ion mobility-mass spectrometry (IM-MS)
Aβ42 was dissolved in 0.1% NH4OH at 400 μM, and DDC was solubilized in EtOH at 8 mM, followed by a 10-fold and 100-fold dilution with 25 mM ammonium acetate (pH 7.4), respectively. The resultant solution (Aβ42: 40 μM, DDC: 80 μM) was centrifuged for 4 min at 2000g (4 °C) before infusion into the MS apparatus using a glass capillary (Nanoflow Probe Tip, Waters). Mass spectra and ion mobility experiments were accomplished on SYNAPT G2-Si HDMS (Waters) using a nanoelectrospray as an ionization source (NanoESI-TOF-MS). The instrument was operated in negative ion mode with a capillary voltage of 1.0 kV, a sample cone voltage of 10 V, a source temperature of 50 °C, and a desolvation temperature of 50 °C. For the ion mobility measurement, nitrogen gas was used in the ion mobility cell, and the cell pressure was maintained at approximately 2.95 mbar with a wave velocity of 300–1000 m s−1 and a wave height of 10–40 V. Data acquisition and processing were performed with the MassLynx (V4.1) and DriftScope (V2.8) software supplied with the instrument. The CsI cluster ions were used for m/z scale as a calibrator.Animal experiments
All experimental procedures were performed in accordance with specified guidelines for the care and use of laboratory animals, and were approved by the Animal Care and Use Committee of Chiba University. Tg2576/PS2 mice,55 which were developed by crossbreeding with Tg2576 and human mutant PS2 (N141I) transgenic mice, at 9 months of age and APPNL-G-F/NL-G-F knock-in mice58 at 26 months of age were used for experiments. All mice were maintained in a 24 ± 1 °C room, with 55% ± 10% relative humidity, under a 12h light/dark cycle, with ad libitum access to food and drinking water. DDC (3 mg) suspended in 0.3 mL polypropyleneglycol was orally administered to the mice starved overnight. Mice as a control received administration of polypropyleneglycol without DDC. Blood, brain, and small intestine were collected under anesthesia by a combination of anesthetics (medetomidine [0.3 mg kg−1], midazolam [4.0 mg kg−1], and butorphanol [5.0 mg kg−1]) after 30 min of DDC administration. Blood was put in the tube coated with EDTA-2Na, and then centrifuged for 5 min at 6000 rpm at room temperature to obtain plasma. Brain and small intestine were harvested after perfusion of ice-cold PBS from the heart to completely remove the blood. Brain, small intestine, and plasma were stored at −80 °C until use.
Immunoaffinity purification mass spectrometry (IP-MS)
For immunoprecipitation of Aβ from brain lysate of mice, we added 25 μg of anti-N-terminus of Aβ antibody (82E1, 1 mg mL−1, IBL) to brain lysate (∼6 mg protein), followed by incubation on a rotating platform for 1 h at 23 °C. Brain lysate was prepared using Tris–HCl buffer without detergents to avoid hampering the interaction of Aβ-ligand, as previously reported.84 After centrifugation for 20 min at 17900g at 4 °C, the supernatant was removed, and the precipitate was washed with 0.9% NaCl (500 μL) twice and sequentially washed with distilled water (500 μL) twice. After the final wash, the precipitate was dried in vacuo, and stored at −80 °C until use.
For mass analysis of immunoprecipitated Aβ, the dried precipitate was dissolved with 10 μL of 50% acetonitrile containing 0.1% trifluoroacetic acid, and 0.5 μL of the solution was applied directly onto a MALDI-target plate. The droplet was mixed on the spot with 0.5 μL of matrix solution of saturated α-cyano 4-hydroxy cinnaminic acid (CHCA, Sigma) or sinapic acid (Sigma) dissolved in 50% acetonitrile, 0.1% trifluoroacetic acid, and allowed to dry. Mass spectra were acquired on a Voyager DE Pro (Applied Biosystems, Foster City, CA, USA) in the positive ion linear mode. External calibration was done with respect to synthetic standard of Aβ42,81,82 E22G-Aβ42,83 insulin (Sigma), and insulin β oxidized form (Sigma).
Statistical analysis
The statistical significance of differences was analysed by one-way analysis of variance and post hoc multiple comparisons using Tukey's test. Statistical significance was defined as P < 0.05.Author contributions
K. M., and K. I. designed the research. K. M., Y. S., K. T. and M. H. performed in vitro experiments. T. K. and K. H. performed IM-MS experiments. N. I. performed in vivo experiments under the guidance of T. S., K. M. and K. I. analyzed data. K. M. wrote the manuscript. K. I. edited the manuscript.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This study was supported in part by JSPS KAKENHI Grant 16H06194 to K. M., 26221202 to K. I. We thank Drs Takaomi C. Saido and Takashi Saito (RIKEN Brain Science Institute) for providing AppNL-G-F/NL-G-F mice, Drs Junji Sugiyama and Tatsuya Awano (Graduate School of Agriculture, Kyoto University) for support of TEM measurement, Dr Takeshi Ara (Graduate School of Agriculture, Kyoto University) for support of FlavonoidSearch, and Dr Youhei Miyanoiri (Institute for Protein Research, Osaka University) for support of SOFAST-HMQC measurement.References
- G. G. Glenner and C. W. Wong, Alzheimer's disease: initial report of the purification and characterization of a novel cerebrovascular amyloid protein, Biochem. Biophys. Res. Commun., 1984, 120, 885–890 CrossRef CAS PubMed.
- C. L. Masters, G. Simms, N. A. Weinman, G. Multhaup, B. L. McDonald and K. Beyreuther, Amyloid plaque core protein in Alzheimer disease and Down syndrome, Proc. Natl. Acad. Sci. U. S. A., 1985, 82, 4245–4249 CrossRef CAS PubMed.
- R. Roychaudhuri, M. Yang, M. M. Hoshi and D. B. Teplow, Amyloid β-protein assembly and Alzheimer disease, J. Biol. Chem., 2009, 284, 4749–4753 CrossRef CAS PubMed.
- I. Benilova, E. Karran and B. De Strooper, The toxic Aβ oligomer and Alzheimer's disease: an emperor in need of clothes, Nat. Neurosci., 2012, 15, 349–357 CrossRef CAS PubMed.
- K. Hasegawa, I. Yamaguchi, S. Omata, F. Gejyo and H. Naiki, Interaction between Aβ(1-42) and Aβ(1-40) in Alzheimer's β-amyloid fibril formation in vitro, Biochemistry, 1999, 38, 15514–15521 CrossRef CAS PubMed.
- E. Hellstrand, B. Boland, D. M. Walsh and S. Linse, Amyloid β-protein aggregation produces highly reproducible kinetic data and occurs by a two-phase process, ACS Chem. Neurosci., 2010, 1, 13–18 CrossRef CAS PubMed.
- I. M. Ilie and A. Caflisch, Simulation studies of amyloidogenic polypeptides and their aggregates, Chem. Rev., 2019, 119, 6956–6993 CrossRef CAS PubMed.
- D. J. Selkoe and J. Hardy, The amyloid hypothesis of Alzheimer's disease at 25 years, EMBO Mol. Med., 2016, 8, 595–608 CrossRef CAS PubMed.
- E. N. Cline, M. A. Bicca, K. L. Viola and W. L. Klein, The amyloid- oligomer hypothesis: beginning of the third decade, J. Alzheimer's Dis., 2018, 64, S567–S610 CAS.
- J. Bieschke, M. Herbst, T. Wiglenda, R. P. Friedrich, A. Boeddrich, F. Schiele, D. Kleckers, J. M. Lopez del Amo, B. A. Gruning, Q. Wang, M. R. Schmidt, R. Lurz, R. Anwyl, S. Schnoegl, M. Fandrich, R. F. Frank, B. Reif, S. Gunther, D. M. Walsh and E. E. Wanker, Small-molecule conversion of toxic oligomers to nontoxic β-sheet-rich amyloid fibrils, Nat. Chem. Biol., 2011, 8, 93–101 CrossRef PubMed.
- K. Murakami, S. Horii, M. Hanaki and K. Irie, Searching for natural products That delay nucleation phase and promote elongation phase of amyloid β42 toward Alzheimer's disease therapeutics, ACS Chem. Neurosci., 2021, 12, 3467–3476 CrossRef CAS PubMed.
- G. Yamin, K. Ono, M. Inayathullah and D. B. Teplow, Amyloid β-protein assembly as a therapeutic target of Alzheimer's disease, Curr. Pharm. Des., 2008, 14, 3231–3246 CrossRef CAS PubMed.
- P. Williams, A. Sorribas and M. J. Howes, Natural products as a source of Alzheimer's drug leads, Nat. Prod. Rep., 2011, 28, 48–77 RSC.
- S. J. Lee, E. Nam, H. J. Lee, M. G. Savelieff and M. H. Lim, Towards an understanding of amyloid-β oligomers: characterization, toxicity mechanisms, and inhibitors, Chem. Soc. Rev., 2017, 46, 310–323 RSC.
- K. Murakami and K. Irie, Three structural features of functional food components and herbal medicine with amyloid β42 anti-aggregation properties, Molecules, 2019, 24, 2125 CrossRef CAS PubMed.
- M. Sato, K. Murakami, M. Uno, Y. Nakagawa, S. Katayama, K. Akagi, Y. Masuda, K. Takegoshi and K. Irie, Site-specific inhibitory mechanism for amyloid β42 aggregation by catechol-type flavonoids targeting the Lys residues, J. Biol. Chem., 2013, 288, 23212–23224 CrossRef CAS PubMed.
- M. Hanaki, K. Murakami, K. Akagi and K. Irie, Structural insights into mechanisms for inhibiting amyloid β42 aggregation by non-catechol-type flavonoids, Bioorg. Med. Chem., 2016, 24, 304–313 CrossRef CAS PubMed.
- M. Hanaki, K. Murakami, S. Katayama, K. I. Akagi and K. Irie, Mechanistic analyses of the suppression of amyloid β42 aggregation by apomorphine, Bioorg. Med. Chem., 2018, 26, 1538–1546 CrossRef CAS PubMed.
- K. Murakami, T. Yoshioka, S. Horii, M. Hanaki, S. Midorikawa, S. Taniwaki, H. Gunji, K. I. Akagi, T. Kawase, K. Hirose and K. Irie, Role of the carboxy groups of triterpenoids in their inhibition of the nucleation of amyloid β42 required for forming toxic oligomers, Chem. Commun., 2018, 54, 6272–6275 RSC.
- K. Tsukada, S. Shinki, A. Kaneko, K. Murakami, K. Irie, M. Murai, H. Miyoshi, S. Dan, K. Kawaji, H. Hayashi, E. N. Kodama, A. Hori, E. Salim, T. Kuraishi, N. Hirata, Y. Kanda and T. Asai, Synthetic biology based construction of biological activity-related library of fungal decalin-containing diterpenoid pyrones, Nat. Commun., 2020, 11, 1830 CrossRef CAS PubMed.
- Y. Morishita, K. Tsukada, K. Murakami, K. Irie and T. Asai, Synthetic biology-based discovery of diterpenoid pyrones from the genome of Eupenicillium shearii, J. Nat. Prod., 2022, 85, 384–390 CrossRef CAS PubMed.
- K. Ono, K. Hasegawa, H. Naiki and M. Yamada, Curcumin has potent anti-amyloidogenic effects for Alzheimer's β-amyloid fibrils in vitro, J. Neurosci. Res., 2004, 75, 742–750 CrossRef CAS PubMed.
- F. Yang, G. P. Lim, A. N. Begum, O. J. Ubeda, M. R. Simmons, S. S. Ambegaokar, P. P. Chen, R. Kayed, C. G. Glabe, S. A. Frautschy and G. M. Cole, Curcumin inhibits formation of amyloid β oligomers and fibrils, binds plaques, and reduces amyloid in vivo, J. Biol. Chem., 2005, 280, 5892–5901 CrossRef CAS PubMed.
- Y. J. Wang, M. H. Pan, A. L. Cheng, L. I. Lin, Y. S. Ho, C. Y. Hsieh and J. K. Lin, Stability of curcumin in buffer solutions and characterization of its degradation products, J. Pharm. Biomed. Anal., 1997, 15, 1867–1876 CrossRef CAS PubMed.
- H. H. Tonnesen, J. Karlsen and G. B. van Henegouwen, Studies on curcumin and curcuminoids. VIII. Photochemical stability of curcumin, Z. Lebensm. -Unters. Forsch., 1986, 183, 116–122 CrossRef CAS PubMed.
- M. Griesser, V. Pistis, T. Suzuki, N. Tejera, D. A. Pratt and C. Schneider, Autoxidative and cyclooxygenase-2 catalyzed transformation of the dietary chemopreventive agent curcumin, J. Biol. Chem., 2011, 286, 1114–1124 CrossRef CAS PubMed.
- K. M. Nelson, J. L. Dahlin, J. Bisson, J. Graham, G. F. Pauli and M. A. Walters, The essential medicinal chemistry of curcumin, J. Med. Chem., 2017, 60, 1620–1637 CrossRef CAS PubMed.
- Y. Izumi, A. Matsumura, S. Wakita, K. Akagi, H. Fukuda, T. Kume, K. Irie, Y. Takada-Takatori, H. Sugimoto, T. Hashimoto and A. Akaike, Isolation, identification, and biological evaluation of Nrf2-ARE activator from the leaves of green perilla (Perilla frutescens var. crispa f. viridis), Free Radical Biol. Med., 2012, 53, 669–679 CrossRef CAS PubMed.
- M. Iwasaki, N. Izuo, Y. Izumi, Y. Takada-Takatori, A. Akaike and T. Kume, Protective effect of green perilla-derived chalcone derivative DDC on amyloid β protein-induced neurotoxicity in primary cortical neurons, Biol. Pharm. Bull., 2019, 42, 1942–1946 CrossRef CAS PubMed.
- K. Ichino, H. Tanaka, K. Ito, T. Tanaka and M. Mizuno, Synthesis of helilandin B, pashanone, and their isomers, J. Nat. Prod., 1988, 51, 906–914 CrossRef CAS PubMed.
- A. Aliyan, N. P. Cook and A. A. Marti, Interrogating amyloid aggregates using fluorescent probes, Chem. Rev., 2019, 119, 11819–11856 CrossRef CAS PubMed.
- Y. Suzuki, J. R. Brender, K. Hartman, A. Ramamoorthy and E. N. Marsh, Alternative pathways of human islet amyloid polypeptide aggregation distinguished by (19)f nuclear magnetic resonance-detected kinetics of monomer consumption, Biochemistry, 2012, 51, 8154–8162 CrossRef CAS PubMed.
- S. A. Hudson, H. Ecroyd, T. W. Kee and J. A. Carver, The thioflavin T fluorescence assay for amyloid fibril detection can be biased by the presence of exogenous compounds, FEBS J., 2009, 276, 5960–5972 CrossRef CAS PubMed.
- B. M. Muller, J. Mai, R. A. Yocum and M. J. Adler, Impact of mono- and disubstitution on the colorimetric dynamic covalent switching chalcone/flavanone scaffold, Org. Biomol. Chem., 2014, 12, 5108–5114 RSC.
- N. Akimoto, T. Ara, D. Nakajima, K. Suda, C. Ikeda, S. Takahashi, R. Muneto, M. Yamada, H. Suzuki, D. Shibata and N. Sakurai, FlavonoidSearch: A system for comprehensive flavonoid annotation by mass spectrometry, Sci. Rep., 2017, 7, 1243 CrossRef PubMed.
- F. Allen, A. Pon, M. Wilson, R. Greiner and D. Wishart, CFM-ID: a web server for annotation, spectrum prediction and metabolite identification from tandem mass spectra, Nucleic Acids Res., 2014, 42, W94–W99 CrossRef CAS.
- C. Scheele, E. Wollenweber and F. J. Arriaga-Giner, New flavonoids from cheilanthoid ferns, J. Nat. Prod., 1987, 50, 181–187 CrossRef CAS.
- A. Nakajima, Y. Yamamoto, N. Yoshinaka, M. Namba, H. Matsuo, T. Okuyama, E. Yoshigai, T. Okumura, M. Nishizawa and Y. Ikeya, A new flavanone and other flavonoids from green perilla leaf extract inhibit nitric oxide production in interleukin 1β-treated hepatocytes, Biosci., Biotechnol., Biochem., 2015, 79, 138–146 CrossRef CAS PubMed.
- R. Kayed, E. Head, J. L. Thompson, T. M. McIntire, S. C. Milton, C. W. Cotman and C. G. Glabe, Common structure of soluble amyloid oligomers implies common mechanism of pathogenesis, Science, 2003, 300, 486–489 CrossRef CAS PubMed.
- P. Schanda and B. Brutscher, Very fast two-dimensional NMR spectroscopy for real-time investigation of dynamic events in proteins on the time scale of seconds, J. Am. Chem. Soc., 2005, 127, 8014–8015 CrossRef CAS PubMed.
- L. Hou, H. Shao, Y. Zhang, H. Li, N. K. Menon, E. B. Neuhaus, J. M. Brewer, I. J. Byeon, D. G. Ray, M. P. Vitek, T. Iwashita, R. A. Makula, A. B. Przybyla and M. G. Zagorski, Solution NMR studies of the Aβ(1-40) and Aβ(1-42) peptides establish that the Met35 oxidation state affects the mechanism of amyloid formation, J. Am. Chem. Soc., 2004, 126, 1992–2005 CrossRef CAS PubMed.
- R. P. Friedland, J. M. Tedesco, A. C. Wilson, C. S. Atwood, M. A. Smith, G. Perry and M. G. Zagorski, Antibodies to potato virus Y bind the amyloid β peptide: immunohistochemical and NMR studies, J. Biol. Chem., 2008, 283, 22550–22556 CrossRef CAS PubMed.
- D. K. Weber, M. A. Sani and J. D. Gehman, A routine method for cloning, expressing and purifying Aβ(1-42) for structural NMR studies, Amino Acids, 2014, 46, 2415–2426 CrossRef CAS PubMed.
- M. P. Williamson, Y. Suzuki, N. T. Bourne and T. Asakura, Binding of amyloid β-peptide to ganglioside micelles is dependent on histidine-13, Biochem. J., 2006, 397, 483–490 CrossRef CAS PubMed.
- Y. Xiao, B. Ma, D. McElheny, S. Parthasarathy, F. Long, M. Hoshi, R. Nussinov and Y. Ishii, Aβ(1-42) fibril structure illuminates self-recognition and replication of amyloid in Alzheimer's disease, Nat. Struct. Mol. Biol., 2015, 22, 499–505 CrossRef CAS PubMed.
- M. T. Colvin, R. Silvers, Q. Z. Ni, T. V. Can, I. Sergeyev, M. Rosay, K. J. Donovan, B. Michael, J. Wall, S. Linse and R. G. Griffin, Atomic resolution structure of monomorphic Aβ42 amyloid fibrils, J. Am. Chem. Soc., 2016, 138, 9663–9674 CrossRef CAS PubMed.
- M. A. Walti, F. Ravotti, H. Arai, C. G. Glabe, J. S. Wall, A. Bockmann, P. Guntert, B. H. Meier and R. Riek, Atomic-resolution structure of a disease-relevant Aβ(1-42) amyloid fibril, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, E4976–E4984 CrossRef CAS PubMed.
- Y. Yang, D. Arseni, W. Zhang, M. Huang, S. Lovestam, M. Schweighauser, A. Kotecha, A. G. Murzin, S. Y. Peak-Chew, J. Macdonald, I. Lavenir, H. J. Garringer, E. Gelpi, K. L. Newell, G. G. Kovacs, R. Vidal, B. Ghetti, B. Ryskeldi-Falcon, S. H. W. Scheres and M. Goedert, Cryo-EM structures of amyloid-β 42 filaments from human brains, Science, 2022, 375, 167–172 CrossRef CAS PubMed.
- M. Ahmed, J. Davis, D. Aucoin, T. Sato, S. Ahuja, S. Aimoto, J. I. Elliott, W. E. Van Nostrand and S. O. Smith, Structural conversion of neurotoxic amyloid-β(1-42) oligomers to fibrils, Nat. Struct. Mol. Biol., 2010, 17, 561–567 CrossRef CAS PubMed.
- K. J. Barnham, F. Haeffner, G. D. Ciccotosto, C. C. Curtain, D. Tew, C. Mavros, K. Beyreuther, D. Carrington, C. L. Masters, R. A. Cherny, R. Cappai and A. I. Bush, Tyrosine gated electron transfer is key to the toxic mechanism of Alzheimer's disease β-amyloid, FASEB J., 2004, 18, 1427–1429 CrossRef CAS PubMed.
- K. Murakami, K. Irie, H. Ohigashi, H. Hara, M. Nagao, T. Shimizu and T. Shirasawa, Formation and stabilization model of the 42-mer Aβ radical: implications for the long-lasting oxidative stress in Alzheimer's disease, J. Am. Chem. Soc., 2005, 127, 15168–15174 CrossRef CAS PubMed.
- K. Murakami, M. Tokuda, T. Suzuki, Y. Irie, M. Hanaki, N. Izuo, Y. Monobe, K. Akagi, R. Ishii, H. Tatebe, T. Tokuda, M. Maeda, T. Kume, T. Shimizu and K. Irie, Monoclonal antibody with conformational specificity for a toxic conformer of amyloid β42 and its application toward the Alzheimer's disease diagnosis, Sci. Rep., 2016, 6, 29038 CrossRef CAS PubMed.
- W. Hoffmann, G. von Helden and K. Pagel, Ion mobility-mass spectrometry and orthogonal gas-phase techniques to study amyloid formation and inhibition, Curr. Opin. Struct. Biol., 2017, 46, 7–15 CrossRef CAS PubMed.
- H. M. Britt, T. Cragnolini and K. Thalassinos, Integration of mass spectrometry data for structural biology, Chem. Rev., 2022, 122, 7952–7986 CrossRef CAS PubMed.
- T. Toda, Y. Noda, G. Ito, M. Maeda and T. Shimizu, Presenilin-2 mutation causes early amyloid accumulation and memory impairment in a transgenic mouse model of Alzheimer's disease, J. Biomed. Biotechnol., 2011, 2011, 617974 Search PubMed.
- T. A. Lanz, K. M. Wood, K. E. Richter, C. E. Nolan, S. L. Becker, N. Pozdnyakov, B. A. Martin, P. Du, C. E. Oborski, D. E. Wood, T. M. Brown, J. E. Finley, S. A. Sokolowski, C. D. Hicks, K. J. Coffman, K. F. Geoghegan, M. A. Brodney, D. Liston and B. Tate, Pharmacodynamics and pharmacokinetics of the γ-secretase inhibitor PF-3084014, J. Pharmacol. Exp. Ther., 2010, 334, 269–277 CrossRef CAS PubMed.
- Y. Hori, T. Hashimoto, Y. Wakutani, K. Urakami, K. Nakashima, M. M. Condron, S. Tsubuki, T. C. Saido, D. B. Teplow and T. Iwatsubo, The Tottori (D7N) and English (H6R) familial Alzheimer disease mutations accelerate Aβ fibril formation without increasing protofibril formation, J. Biol. Chem., 2007, 282, 4916–4923 CrossRef CAS PubMed.
- T. Saito, Y. Matsuba, N. Mihira, J. Takano, P. Nilsson, S. Itohara, N. Iwata and T. C. Saido, Single App knock-in mouse models of Alzheimer's disease, Nat. Neurosci., 2014, 17, 661–663 CrossRef CAS PubMed.
- K. M. Nelson, J. L. Dahlin, J. Bisson, J. Graham, G. F. Pauli and M. A. Walters, Curcumin may (not) defy science, ACS Med. Chem. Lett., 2017, 8, 467–470 CrossRef CAS PubMed.
- L. Abranko and B. Szilvassy, Mass spectrometric profiling of flavonoid glycoconjugates possessing isomeric aglycones, J. Mass Spectrom., 2015, 50, 71–80 CrossRef CAS PubMed.
- Z. Yan, G. Lin, Y. Ye, Y. Wang and R. Yan, A generic multiple reaction monitoring based approach for plant flavonoids profiling using a triple quadrupole linear ion trap mass spectrometry, J. Am. Soc. Mass Spectrom., 2014, 25, 955–965 CrossRef CAS PubMed.
- A. Lord, H. Englund, L. Soderberg, S. Tucker, F. Clausen, L. Hillered, M. Gordon, D. Morgan, L. Lannfelt, F. E. Pettersson and L. N. Nilsson, Amyloid-β protofibril levels correlate with spatial learning in Arctic Alzheimer's disease transgenic mice, FEBS J., 2009, 276, 995–1006 CrossRef CAS PubMed.
- L. Ho, M. G. Ferruzzi, E. M. Janle, J. Wang, B. Gong, T. Y. Chen, J. Lobo, B. Cooper, Q. L. Wu, S. T. Talcott, S. S. Percival, J. E. Simon and G. M. Pasinetti, Identification of brain-targeted bioactive dietary quercetin-3-O-glucuronide as a novel intervention for Alzheimer's disease, FASEB J., 2013, 27, 769–781 CrossRef CAS PubMed.
- J. Wang, M. G. Ferruzzi, L. Ho, J. Blount, E. M. Janle, B. Gong, Y. Pan, G. A. Gowda, D. Raftery, I. Arrieta-Cruz, V. Sharma, B. Cooper, J. Lobo, J. E. Simon, C. Zhang, A. Cheng, X. Qian, K. Ono, D. B. Teplow, C. Pavlides, R. A. Dixon and G. M. Pasinetti, Brain-targeted proanthocyanidin metabolites for Alzheimer's disease treatment, J. Neurosci., 2012, 32, 5144–5150 CrossRef CAS PubMed.
- D. A. Nation, M. D. Sweeney, A. Montagne, A. P. Sagare, L. M. D'Orazio, M. Pachicano, F. Sepehrband, A. R. Nelson, D. P. Buennagel, M. G. Harrington, T. L. S. Benzinger, A. M. Fagan, J. M. Ringman, L. S. Schneider, J. C. Morris, H. C. Chui, M. Law, A. W. Toga and B. V. Zlokovic, Blood-brain barrier breakdown is an early biomarker of human cognitive dysfunction, Nat. Med., 2019, 25, 270–276 CrossRef CAS PubMed.
- S. Pype, D. Moechars, L. Dillen and M. Mercken, Characterization of amyloid β peptides from brain extracts of transgenic mice overexpressing the London mutant of human amyloid precursor protein, J. Neurochem., 2003, 84, 602–609 CrossRef CAS PubMed.
- J. G. Sheng, S. H. Bora, G. Xu, D. R. Borchelt, D. L. Price and V. E. Koliatsos, Lipopolysaccharide-induced-neuroinflammation increases intracellular accumulation of amyloid precursor protein and amyloid β peptide in APPswe transgenic mice, Neurobiol. Dis., 2003, 14, 133–145 CrossRef CAS PubMed.
- P. Velander, L. Wu, F. Henderson, S. Zhang, D. R. Bevan and B. Xu, Natural product-based amyloid inhibitors, Biochem. Pharmacol., 2017, 139, 40–55 CrossRef CAS PubMed.
- M. Zhu, S. Rajamani, J. Kaylor, S. Han, F. Zhou and A. L. Fink, The flavonoid baicalein inhibits fibrillation of α-synuclein and disaggregates existing fibrils, J. Biol. Chem., 2004, 279, 26846–26857 CrossRef CAS PubMed.
- F. L. Palhano, J. Lee, N. P. Grimster and J. W. Kelly, Toward the molecular mechanism(s) by which EGCG treatment remodels mature amyloid fibrils, J. Am. Chem. Soc., 2013, 135, 7503–7510 CrossRef CAS PubMed.
- P. Banerjee, A. Sahoo, S. Anand, A. Ganguly, G. Righi, P. Bovicelli, L. Saso and S. Chakrabarti, Multiple mechanisms of iron-induced amyloid β-peptide accumulation in SHSY5Y cells: protective action of negletein, NeuroMol. Med., 2014, 16, 787–798 CrossRef CAS PubMed.
- J. L. Wolfender, M. Litaudon, D. Touboul and E. F. Queiroz, Innovative omics-based approaches for prioritisation and targeted isolation of natural products - new strategies for drug discovery, Nat. Prod. Rep., 2019, 36, 855–868 RSC.
- A. Stavrianidi, A classification of liquid chromatography mass spectrometry techniques for evaluation of chemical composition and quality control of traditional medicines, J. Chromatogr. A, 2020, 1609, 460501 CrossRef CAS PubMed.
- T. Tsujimoto, R. Arai, T. Yoshitomi, Y. Yamamoto, Y. Ozeki, T. Hakamatsuka and N. Uchiyama, UHPLC/MS and NMR-based metabolomic analysis of dried water extract of citrus-type crude drugs, Chem. Pharm. Bull., 2021, 69, 741–746 CrossRef CAS PubMed.
- M. Hanaki, K. Murakami, H. Gunji and K. Irie, Activity-differential search for amyloid-β aggregation inhibitors using LC-MS combined with principal component analysis, Bioorg. Med. Chem. Lett., 2022, 61, 128613 CrossRef CAS PubMed.
- S. Saito, Y. Yamamoto, T. Maki, Y. Hattori, H. Ito, K. Mizuno, M. Harada-Shiba, R. N. Kalaria, M. Fukushima, R. Takahashi and M. Ihara, Taxifolin inhibits amyloid-β oligomer formation and fully restores vascular integrity and memory in cerebral amyloid angiopathy, Acta Neuropathol. Commun., 2017, 5, 26 CrossRef PubMed.
- T. Inoue, S. Saito, M. Tanaka, H. Yamakage, T. Kusakabe, A. Shimatsu, M. Ihara and N. Satoh-Asahara, Pleiotropic neuroprotective effects of taxifolin in cerebral amyloid angiopathy, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 10031–10038 CrossRef CAS PubMed.
- S. Saito, M. Tanaka, N. Satoh-Asahara, R. O. Carare and M. Ihara, Taxifolin: A potential therapeutic agent for cerebral amyloid angiopathy, Front. Pharmacol., 2021, 12, 643357 CrossRef CAS PubMed.
- V. K. Kolhir, V. A. Bykov, Y. O. Teselkin, I. V. Babenkova, N. A. Tjukavkina, I. A. Rulenko, Y. A. Kolesnik and A. A. Eichholz, Use of a new antioxidant diquertin as an adjuvant in the therapy of patients with acute pneumonia, Phytother. Res., 1998, 12, 606–608 CrossRef CAS.
- D. J. Irwin, V. M. Lee and J. Q. Trojanowski, Parkinson's disease dementia: convergence of α-synuclein, tau and amyloid-β pathologies, Nat. Rev. Neurosci., 2013, 14, 626–636 CrossRef CAS PubMed.
- K. Irie, K. Oie, A. Nakahara, Y. Yanai, H. Ohigashi, P. A. Wender, H. Fukuda, H. Konishi and U. Kikkawa, Molecular basis for protein kinase C isozyme-selective binding: the synthesis, folding, and phorbol ester binding of the cysteine-rich domains of all protein kinase C isozymes, J. Am. Chem. Soc., 1998, 120, 9159–9167 CrossRef CAS.
- H. Fukuda, T. Shimizu, M. Nakajima, H. Mori and T. Shirasawa, Synthesis, aggregation, and neurotoxicity of the Alzheimer's Aβ1-42 amyloid peptide and its isoaspartyl isomers, Bioorg. Med. Chem. Lett., 1999, 9, 953–956 CrossRef CAS PubMed.
- K. Murakami, K. Irie, A. Morimoto, H. Ohigashi, M. Shindo, M. Nagao, T. Shimizu and T. Shirasawa, Neurotoxicity and physicochemical properties of Aβ mutant peptides from cerebral amyloid angiopathy: implication for the pathogenesis of cerebral amyloid angiopathy and Alzheimer's disease, J. Biol. Chem., 2003, 278, 46179–46187 CrossRef CAS PubMed.
- K. Murakami, N. Murata, Y. Noda, S. Tahara, T. Kaneko, N. Kinoshita, H. Hatsuta, S. Murayama, K. J. Barnham, K. Irie, T. Shirasawa and T. Shimizu, SOD1 (Copper/Zinc superoxide dismutase) deficiency drives amyloid β protein oligomerization and memory loss in mouse model of Alzheimer disease, J. Biol. Chem., 2011, 286, 44557–44568 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2cb00194b |
| ‡ Present address: Laboratory of Pharmaceutical Therapy and Neuropharmacology, Faculty of Pharmaceutical Sciences, University of Toyama, Toyama 930-0194, Japan. |
| § Present address: Aging Stress Response Research Project Team, National Center for Geriatrics and Gerontology, Obu 474-8511, Japan. |
| This journal is © The Royal Society of Chemistry 2022 |