
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe covalent reactivity of functionalized 5-hydroxy-butyrolactams is the basis for targeting of fatty acid binding protein 5 (FABP5) by the neurotrophic agent MT-21†
Esben B.
Svenningsen
 a,
Rasmus N.
Ottosen
a,
Katrine H.
Jørgensen
a,
Marija
Nisavic
ab,
Camilla K.
Larsen
c,
Bente K.
Hansen
ad,
Yong
Wang‡
e,
Kresten
Lindorff-Larsen
e,
Thomas
Tørring
c,
Stephan M.
Hacker
f,
Johan
Palmfeldt
b and
Thomas B.
Poulsen
*a
a,
Rasmus N.
Ottosen
a,
Katrine H.
Jørgensen
a,
Marija
Nisavic
ab,
Camilla K.
Larsen
c,
Bente K.
Hansen
ad,
Yong
Wang‡
e,
Kresten
Lindorff-Larsen
e,
Thomas
Tørring
c,
Stephan M.
Hacker
f,
Johan
Palmfeldt
b and
Thomas B.
Poulsen
*a
aDepartment of Chemistry, Aarhus University, DK-8000, Aarhus C, Denmark. E-mail: thpou@chem.au.dk
bDepartment of Clinical Medicine – Research Unit for Molecular Medicine, Aarhus University Hospital, DK-8200 Aarhus N, Denmark
cDepartment of Engineering – Microbial Biosynthesis, Aarhus University, DK-8000 Aarhus C, Denmark
dInterdisciplinary Nanoscience Center (iNANO), Aarhus University, DK-8000 Aarhus C, Denmark
eCopenhagen Biocenter, University of Copenhagen, DK-2200 Copenhagen N, Denmark
fLeiden Institute of Chemistry, Leiden University, NL-2333 CC Leiden, The Netherlands
First published on 7th September 2022
Abstract
Covalently acting compounds experience a strong interest within chemical biology both as molecular probes in studies of fundamental biological mechanisms and/or as novel drug candidates. In this context, the identification of new classes of reactive groups is particularly important as these can expose novel reactivity modes and, consequently, expand the ligandable proteome. Here, we investigated the electrophilic reactivity of the 3-acyl-5-hydroxy-1,5-dihydro-2H-pyrrole-2-one (AHPO) scaffold, a heterocyclic motif that is e.g. present in various bioactive natural products. Our investigations were focused on the compound MT-21 – a simplified structural analogue of the natural product epolactaene – which is known to have both neurotrophic activity and ability to trigger apoptotic cell death. We found that the central N-acyl hemiaminal group of MT-21 can function as an electrophilic centre enabling divergent reactivity with both amine- and thiol-based nucleophiles, which furthermore translated to reactivity with proteins in both cell lysates and live cells. We found that in live cells MT-21 strongly engaged the lipid transport protein fatty acid-binding protein 5 (FABP5) by direct binding to a cysteine residue in the bottom of the ligand binding pocket. Through preparation of a series of MT-21 derivatives, we probed the specificity of this interaction which was found to be strongly dependent on subtle structural changes. Our study suggests that MT-21 may be employed as a tool compound in future studies of the biology of FABP5, which remains incompletely understood. Furthermore, our study has also made clear that other natural products containing the AHPO-motif may likewise possess covalent reactivity and that this property may underlie their biological activity.
Introduction
The rapid expansion of covalent drug discovery1,2 holds substantial promise for the identification of small molecule modulators of an increasingly broad suite of cellular processes and targets, even those previously considered undruggable.3 Additionally, covalent reactivity enables new technologies for the functionalization of native proteins,4–6e.g. to prepare advanced biopharmaceuticals. Thereby, covalent chemistry effectively bridges two major areas within chemical biology: bioconjugation and chemical genetics. Several innovative and instructive examples of covalent chemical biology have emerged recently, including covalent molecular glues7–10 and covalent fragment screening.11–14 These discoveries are enabled in large part by a combination of advances in chemical proteomics,14–16 and an increasing knowledge of the biological performance and characteristics of different classes of reactive groups.17–20 Structural motifs which are non-conventional, or which possess unusual selectivity, are particularly interesting as entry points for new covalent modulator design. Towards that end, natural products provide a valuable resource for continued discovery of novel electrophilic motifs,21–27 but synthetic compounds,28–33 designed or serendipitously discovered,30 are also advancing this area.The 5-hydroxy-butyrolactam core structure34 is prevalent in bioactive compounds and natural products (>100 distinct compounds registered in Natural Product Atlas) and occurs in diverse constellations of peripheral substituents and oxidation states. Within this super-family, the 3-acyl-5-hydroxy-1,5-dihydro-2H-pyrrole-2-one (AHPO) scaffold (Fig. 1a, green highlight) is present in compounds with various bioactivities, such as the platelet aggregation inhibitor PI-091,35,36 the endothelin receptor antagonist oteromycin37 and the potent antibiotic38 and apoptosis inducer39 pyrrocidine A. Despite their enzymatic origin, these natural products are often isolated as diastereomeric mixtures at the N-acyl hemiaminal carbon stereocenter or as mixtures with hydroxy and methoxy substituents. Although these observations can in principle be explained by stereochemical instability during extraction and purification, e.g. using acidic methanol,35 we speculated that this apparent instability might actually be a proxy for latent electrophilic reactivity contained within the AHPO-scaffold. Furthermore, we speculated that such reactivity might, at least in part, determine the biological activities of AHPO-containing compounds. Specifically, we envisioned that reversible iminium ion formation could directly mediate reaction with biological nucleophiles40,41 or, alternatively, the iminium intermediate could undergo tautomerization in situ to form an extended Michael acceptor42 (Fig. 1b). Curiously, we could not find prior studies documenting such reactivity. Consequently, we set out to investigate both the fundamental electrophilic reactivity of the AHPO-scaffold as well as its broader potential in covalent chemical biology. In this manuscript, we report our first studies towards this end.
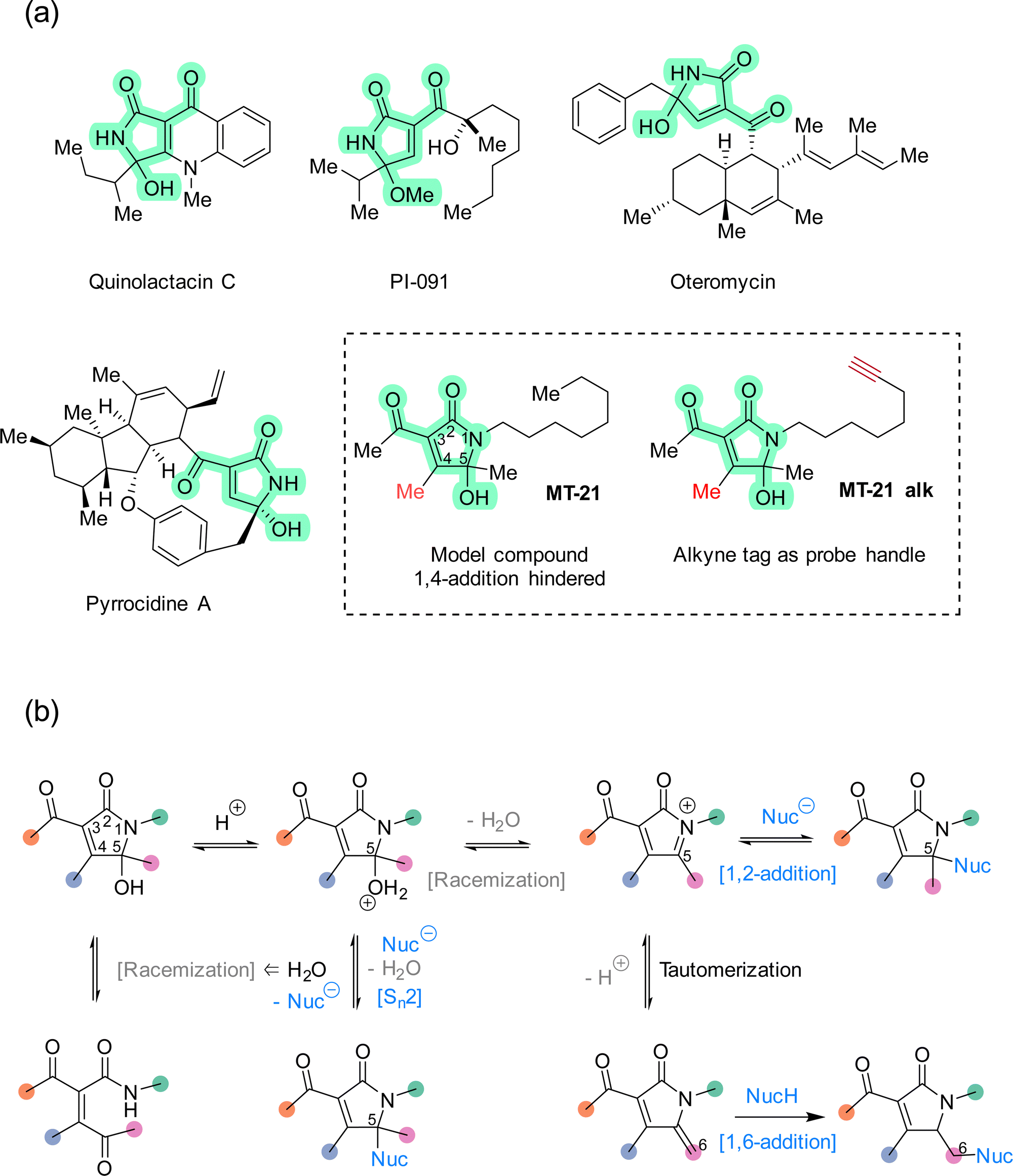 | ||
| Fig. 1 (a) Examples of natural products that contain the AHPO-scaffold and structure of the synthetic compound MT-21 and its associated alkyne-tagged version MT-21 alk. (b) Within the AHPO-scaffold, racemization of the N-acyl hemiaminal stereocenter at C5 may occur either via N-acyl hemiaminal reversal or through iminium ion formation, the latter can further tautomerize to form an extended Michael acceptor. With an AHPO-scaffold sterically blocked at C4, nucleophiles may react at C6 (via 1,6-addition) or at C5 through either 1,2-addition to the iminium ion or by direct substitution. The latter would not compromise stereochemical integrity, unless the nucleophile is water. | ||
We decided to focus our study on the compound MT-21 which contains the central AHPO-scaffold but has the C4-position blocked by a methyl group, effectively hindering another potential electrophilic reaction pathway: 1,4-addition (Fig. 1a). MT-21 was originally developed as a synthetic analogue43 of the neurotrophic natural product epolactaene.44 SH-SY5Y neuroblastoma cells were observed to extend more neurites when treated with the natural product or analogs, including MT-21. Paradoxically, MT-21 has also been shown to induce apoptosis by direct cytochrome C release from mitochondria.45–47 Due to the known involvement of the inner mitochondrial membrane protein ADP/ATP translocase 1 (adenine nucleotide translocase, ANT1) in the permeability transition pore observed in early intrinsic apoptosis, it was speculated whether MT-21 interacted with ANT1. Cytochrome c release mediated by MT-21 was found to be independent of mitochondrial swelling, indicating a permeability transition pore independent mechanism mediated by ANT1 binding. Indeed it was found that MT-21 inhibited ADP uptake in isolated mitochondria and inhibited the interaction between cyclophilin D and ANT1, proposedly through a non-covalent mechanism.47 All in all, the rather divergent observations concerning the biological effects of MT-21 and the absence of a known, common mechanism-of-action – including any indications of involvement of covalent reactivity – makes this compound an interesting AHPO-candidate to study.
Results and discussion
Compound synthesis
We started out our studies by synthesizing MT-21 (see ESI†) for use in reactivity studies. To facilitate later studies in cellular systems or lysates we also prepared an alkyne-tagged analogue, MT-21 alk (Fig. 1a and Fig. 2a) as well as a broader range of alkyne-tagged AHPOs using different synthetic approaches (AHPO-2–8, Fig. 2b and c). Specifically, we employed an alkyne zipper reaction48 to transform oct-3-yn-1-ol (1) to oct-7-yn-1-ol (2) in 96% yield, followed by an Appel reaction and subsequent amine formation by the Gabriel amine synthesis to afford the oct-7-yn-1-amine (4). Next, 4 was coupled with trimethyldioxinone, a source of acetylketene,49 to form β-ketoamide 5 before finally undergoing a Knoevenagel-type aldol condensation with butane-2,3-dione and spontaneous cyclization to form the N-acyl hemiaminal motif in MT-21 alk. In a slightly modified approach, we employed aza-enolate formation from imine 10 to generate the unsymmetric α-diketone 11, which underwent divergent condensation with 3-oxobutanamide to afford AHPO-3 alk and AHPO-4 alk, which could be separated by chromatography. AHPO-derivative AHPO-2 alk, which has the lipid-alkyne chain as part of the acyl functionality attached at C3, was also prepared (Fig. 2c and ESI†). Collectively, the constitutional isomers MT-21 alk, AHPO-2 alk, AHPO-3 alk, and AHPO-4 alk feature subtle differences in the relative attachment of the alkyne-lipid chain and the AHPO-core structure, which we suspected would be of interest in cell-based experiments. Finally, we also prepared a series of MT-21 alk derivatives designed to probe either the effect of minor (Me → Et) substitutions at the putative reactive positions (C4: AHPO-5 alk; C5: AHPO-6 alk) of the AHPO-scaffold or the effect of altering the electron-withdrawing group (CN: AHPO-8 alk; CO2Me: AHPO-7 alk) at C3 (Fig. 2c and ESI†).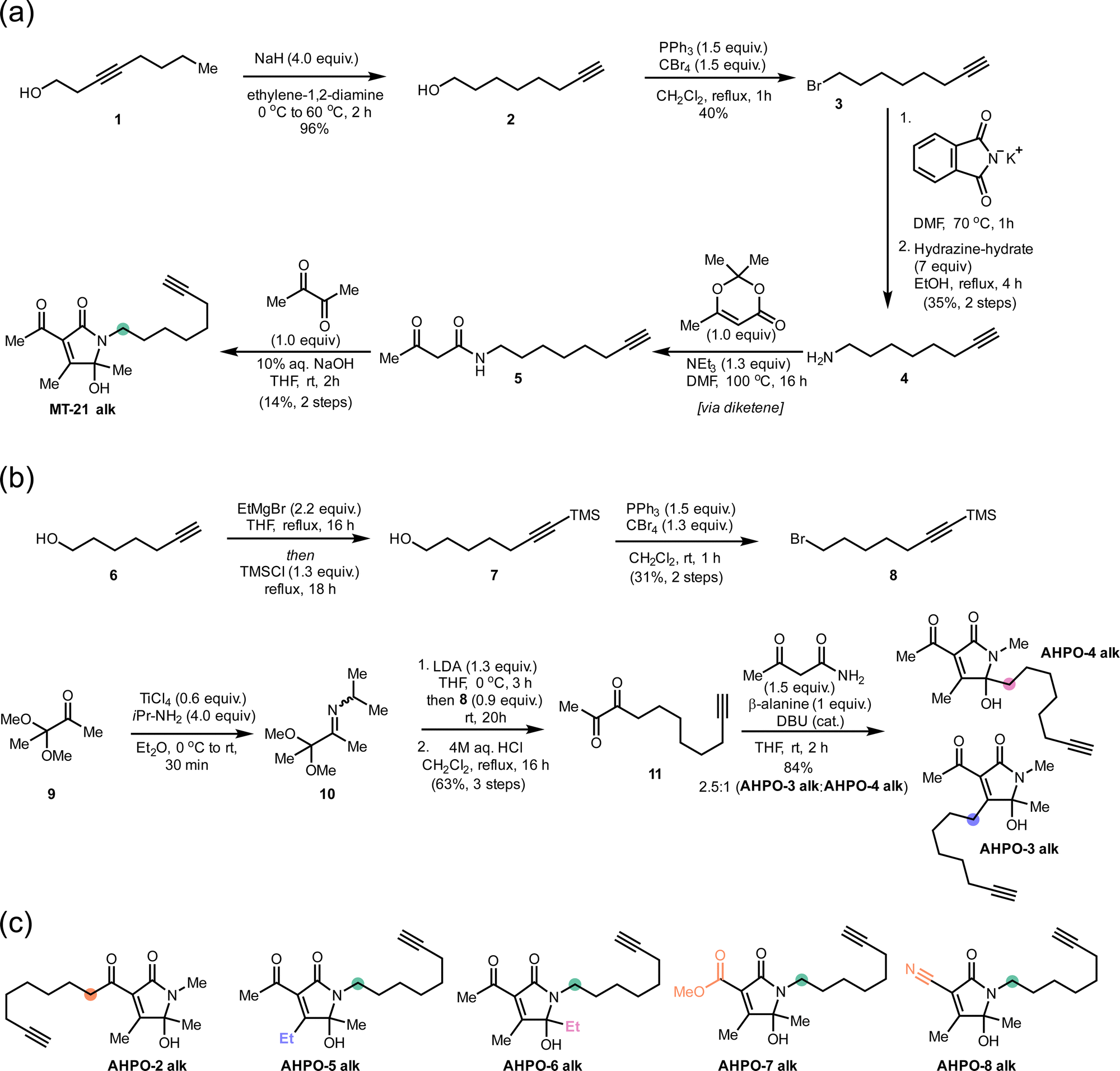 | ||
| Fig. 2 Syntheses of MT-21 alk and various, structurally related AHPO-derivatives. Coloured circles designate the placement of the aliphatic chain (compare to Fig. 1). Other structural modifications compared to MT-21 alk are also indicated by colour-coding on the specific positions that are modified. See ESI† for all details. | ||
Studies of the reactivity of AHPO core towards thiols and amines
To provide the first fundamental information on the intrinsic reactivity of the AHPO-scaffold, we incubated MT-21 with thiol and amine based nucleophiles (Fig. 3a). For the thiol nucleophile, we chose methyl thioglycolate as mimic for proteinogenic cysteine side chains. Initial experiments were performed with 2.5 and 25 equivalents of thiol, in 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 PBS:DMSO at either acidic (pH 4), neutral (pH 7) or basic (pH 10) conditions for four hours and evaluated by HPLC analysis. We observed formation of a single product only at pH 4 (Fig. 3b). Analysis by LC-HRMS revealed a m/z of 370.2047, corresponding to condensation of methyl thioglycolate with MT-21 with loss of one molecule of H2O, which would be consistent with addition at the hemiaminal carbon. To fully elucidate the structure of this compound, the reaction was performed on a preparative scale in CH2Cl2 acidified with TFA. Under these conditions, several products were isolated and – through rigorous NMR spectroscopic analyses (see ESI†) – we identified the major product as conjugate S-Conj1 (Fig. 3a) resulting from apparent thiol-addition to the iminium ion and confirmed its identity with the product formed under aqueous conditions. Additionally, two rather surprising derivatives (S-Conj2 and S-Conj3), in which methyl thioglycolate was connected to C6 were also characterized, with a second thiol unit also being attached at C5 in the case of S-Conj3. The formation of these compounds can be rationalized via an initial 1,6-addition to the enamide derivative (an extended Michael acceptor), followed by oxidation of the resulting enolate50 to reform the AHPO-core (S-Conj2), which can then undergo another addition to form S-Conj3 (Fig. 3b). Interestingly, a related oxidation has been previously reported51 and was recently observed for colibactin.21 Although we did not observe these distinct compounds under aqueous conditions, their formation suggests that the AHPO-scaffold in fact has a rich thiol-reactivity profile. The results also suggest that the AHPO-scaffold is not highly reactive towards thiols (compared with other electrophiles) and that acidic activation/catalysis is required to unlock electrophilic reactivity.
:1 PBS:DMSO at either acidic (pH 4), neutral (pH 7) or basic (pH 10) conditions for four hours and evaluated by HPLC analysis. We observed formation of a single product only at pH 4 (Fig. 3b). Analysis by LC-HRMS revealed a m/z of 370.2047, corresponding to condensation of methyl thioglycolate with MT-21 with loss of one molecule of H2O, which would be consistent with addition at the hemiaminal carbon. To fully elucidate the structure of this compound, the reaction was performed on a preparative scale in CH2Cl2 acidified with TFA. Under these conditions, several products were isolated and – through rigorous NMR spectroscopic analyses (see ESI†) – we identified the major product as conjugate S-Conj1 (Fig. 3a) resulting from apparent thiol-addition to the iminium ion and confirmed its identity with the product formed under aqueous conditions. Additionally, two rather surprising derivatives (S-Conj2 and S-Conj3), in which methyl thioglycolate was connected to C6 were also characterized, with a second thiol unit also being attached at C5 in the case of S-Conj3. The formation of these compounds can be rationalized via an initial 1,6-addition to the enamide derivative (an extended Michael acceptor), followed by oxidation of the resulting enolate50 to reform the AHPO-core (S-Conj2), which can then undergo another addition to form S-Conj3 (Fig. 3b). Interestingly, a related oxidation has been previously reported51 and was recently observed for colibactin.21 Although we did not observe these distinct compounds under aqueous conditions, their formation suggests that the AHPO-scaffold in fact has a rich thiol-reactivity profile. The results also suggest that the AHPO-scaffold is not highly reactive towards thiols (compared with other electrophiles) and that acidic activation/catalysis is required to unlock electrophilic reactivity.
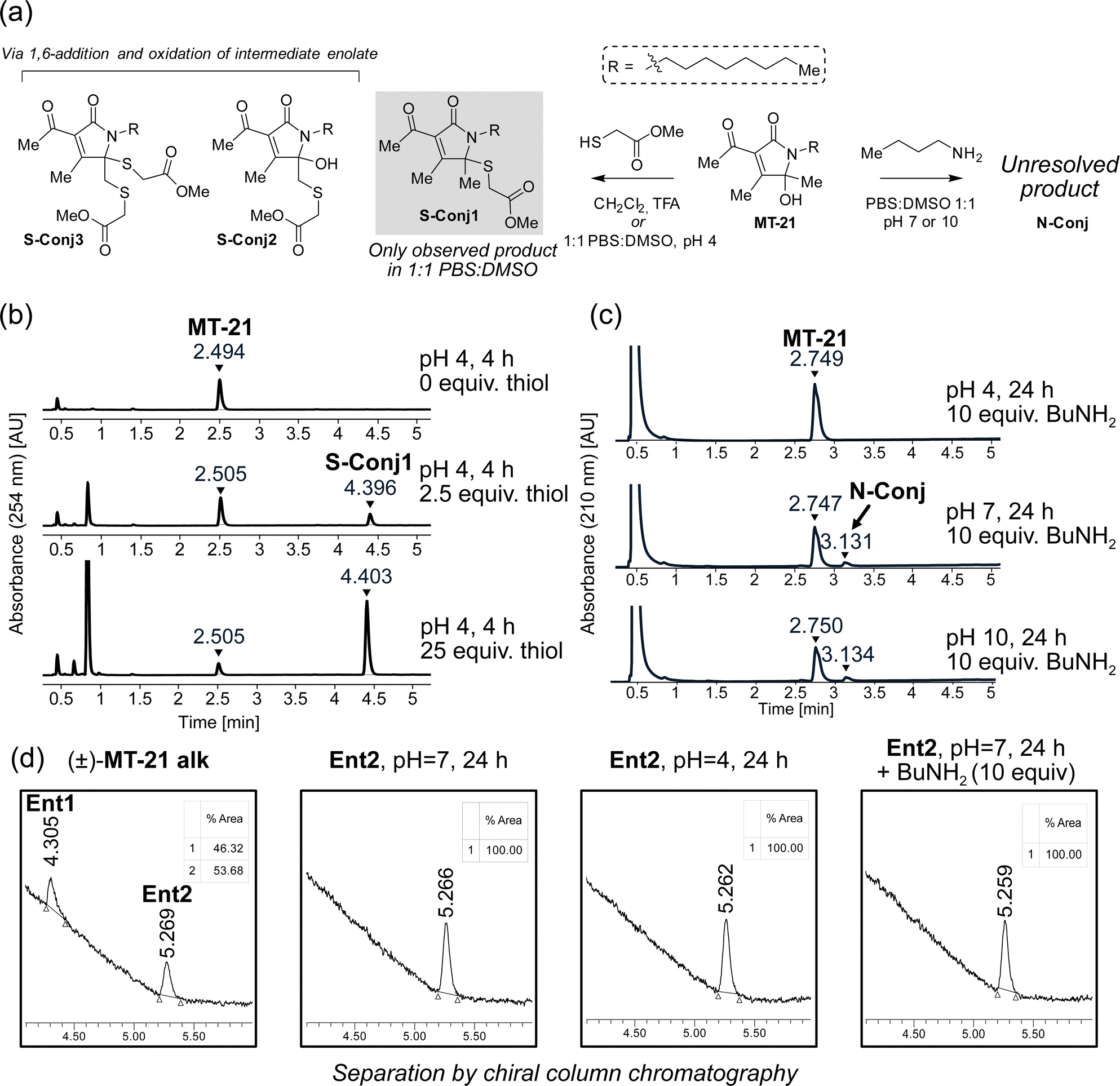 | ||
| Fig. 3 The AHPO-scaffold represented by MT-21 is a dichotomous electrophile. (a) Different conjugation products are formed upon reaction of MT-21 with methyl thioglycolate or n-butylamine acting as simple mimics of biological thiols and amines respectively. (b and c) HPLC-chromatograms show formation of S-Conj1 upon incubation with methyl thioglycolate under acidic conditions (pH = 4) and formation of N-Conj upon incubation with n-butylamine under neutral conditions (pH = 7) in DMSO:PBS 1:1. (d) Studies of the stereochemical stability of MT-21 alk in aqueous conditions. The enantiomers of MT-21 alk were preparatively separated and the stereochemical stability of one of these enantiomers (arbitrarily designated Ent2) was evaluated under the same conditions where conjugation to thiols/amines are observed as shown in (b and c). The stability was evaluated by UPCC chiral chromatography with sCO2/MeCN as mobile phase. | ||
We tested the stability of the conjugate S-Conj1 at acidic, neutral and basic condition. These experiments showed that S-Conj1 has poor stability at basic pH, with concomittant observation of small amounts of parent MT-21 within 30 minutes thereby suggesting that alternative decomposition pathways besides hydrolytic reversal of the conjugation may be operative (Fig. S1, ESI†). At neutral pH the stability was higher, with approximately half of the conjugate lost after 1.5 hours of incubation, while almost full stability was observed at pH 4.
Next, we investigated the reactivity of the scaffold towards simple amine nucleophiles, using butylamine, diethylamine and triethylamine under the same conditions. Interestingly, only butylamine reacted and exclusively at neutral (pH 7) or basic pH (pH 10) (Fig. 3c), forming very minor amounts of a product with m/z 335.2677, which does not match any expected products. The reaction was scaled up in 1:1 PBS:DMSO, pH 7, and the product was purified by reverse phase column chromatography. Despite obtaining full NMR-characterisation (1H-NMR, 13C-NMR, HMBC, HSQC and COSY) we were unable to unequivocally resolve the structure of the product (N-Conj). The NMR-data is available in the ESI.†
Overall, these initial experiments demonstrate that the AHPO-scaffold possesses a range of different electrophilic properties, particularly towards thiols. Nevertheless, they also show rather low reactivity and, therefore, suggests that specific conditions, such as relative positioning of a nucleophilic side chain and other residues, that can act e.g. as general acid catalysts, may be required to favor covalent reactivity of this scaffold. In a similar manner, the resulting thiol-conjugate(s) may also exhibit context-dependent (in)stability.
Stereochemical stability at C5 of the AHPO-scaffold
Our first experiments suggested that an N-acyl iminium-ion may be mediating the reactivity of the AHPO-group towards biological nucleophiles, in particular thiols. In order to study the potential formation of this intermediate under various conditions, we next attempted preparative separation of the two enantiomers of MT-21 alk by chiral HPLC purification using a gradient of hexane/isopropanol on a CHIRALPAK IC column (see Fig. S2, ESI† for HPLC chromatograms and CD spectroscopy). Despite the – potentially – labile nature of the stereocenter at C5 (Fig. 1b), the two enantiomers (Ent1 and Ent2) could be preparatively separated and then subjected to similar conditions as during the conjugation reactions (Fig. 3d). Our rationale for this experiment was as follows: if a rapid equilibrium between the N-acyl hemiaminal and the N-acyliminium-ion exists under the conditions, at which electrophilic reactivity is observed, racemization of the stereocenter should occur. To our surprise, we observed complete stability of the stereocenter both at pH 4 and under incubation with excess butylamine at neutral pH (Fig. 3d), indicating that neither of these conditions lead to rapid imminium-ion formation or, as an alternative racemization pathway, reversible opening of the γ-lactam ring (Fig. 1b). Rather, the results indicate that, at least in the case of the thiol-conjugation product, the reaction occurs through a direct substitution at C5, presumably facilitated by prior or concomittant protonation of the departing hydroxyl group.To test whether the AHPO-scaffold could also be expected to be stereochemically stable during more complex biological experiments (vide infra), we performed a 24 h incubation of one of the purified enantiomers under low-DMSO (2%) conditions in PBS or cell media, in which various Lewis acids are present. In PBS, only very slight racemization (4%) was observed, whereas 14% of the compound racemized in cell media, as indicated by the formation of 7% of the opposite enantiomer (Fig. S2, ESI†).
These findings indicate that a reactive iminium-ion does not rapidly and reversibly form under biologically relevant conditions as might otherwise be expected a priori. Rather, the data suggests that in the complexity of e.g., a proteome, the specific microenvironment surrounding reactive side chains (e.g. cysteines or lysines) may facilitate activation of the AHPO-scaffold with a resulting bias towards certain target sites as the outcome.
MT-21 alk covalently binds proteins in biological contexts
To investigate whether the observed reactivity with thiols and amines could be reconstituted in biological systems we used the alkyne functionalized analog MT-21 alk to probe covalent binding partners in cell lysates and whole cells. The probe was first incubated with the lysates from HCT-116 and MCF-7 cells for 2 hours and the proteins were precipitated with MeOH to remove excess compound. The redissolved proteins were conjugated to TAMRA-N3 by copper catalyzed azide–alkyne cycloaddition (CuAAC) and the proteins were separated by SDS-PAGE. In-gel fluorescence scanning revealed a dose-dependent, but promiscuous, labelling pattern in lysates from both cell lines (Fig. 4a). This experiment clearly demonstrates that the AHPO-group can be expected to act as an covalent modifer in biological systems. In stark contrast to the behaviour in cell lysate, when MT-21 alk was instead incubated in live HCT-116 cells only a few bands were visible (Fig. 4b), most notably an apparent single band around 15 kDa was dose-dependently labelled. We next focussed on the identification of the 15 kDa band that occurred upon labelling with MT-21 alk in cells. Importantly, the labelling could be competed by preincubation with non-functionalized MT-21. In addition to this apparent major binding target of MT-21, other less intense bands could also be discerned. To identify the band at 15 kDa, we treated live HCT-116 cells with MT-21 alk followed by CuAAC with biotin-azide and enrichment on streptavidin beads. The enriched proteins were eluted from the beads by boiling in reducing sample buffer and separated by SDS-PAGE. A single band was visible by Coomassie stain at 15 kDa (Fig. 4c) which was excised from the gel followed by in-gel trypsination and LC-MS/MS analysis of the eluted peptides. This confirmed strong enrichment of fatty acid binding protein 5 (FABP5) and in addition modification of cysteine 120 was observed by MS2-analysis (Fig. S3, ESI†). To confirm the enrichment, we repeated the pulldown experiment and evaluated by western blot, which indeed confirmed that MT-21 alk could covalently bind FABP5 in a MT-21 competitive manner (Fig. 4d).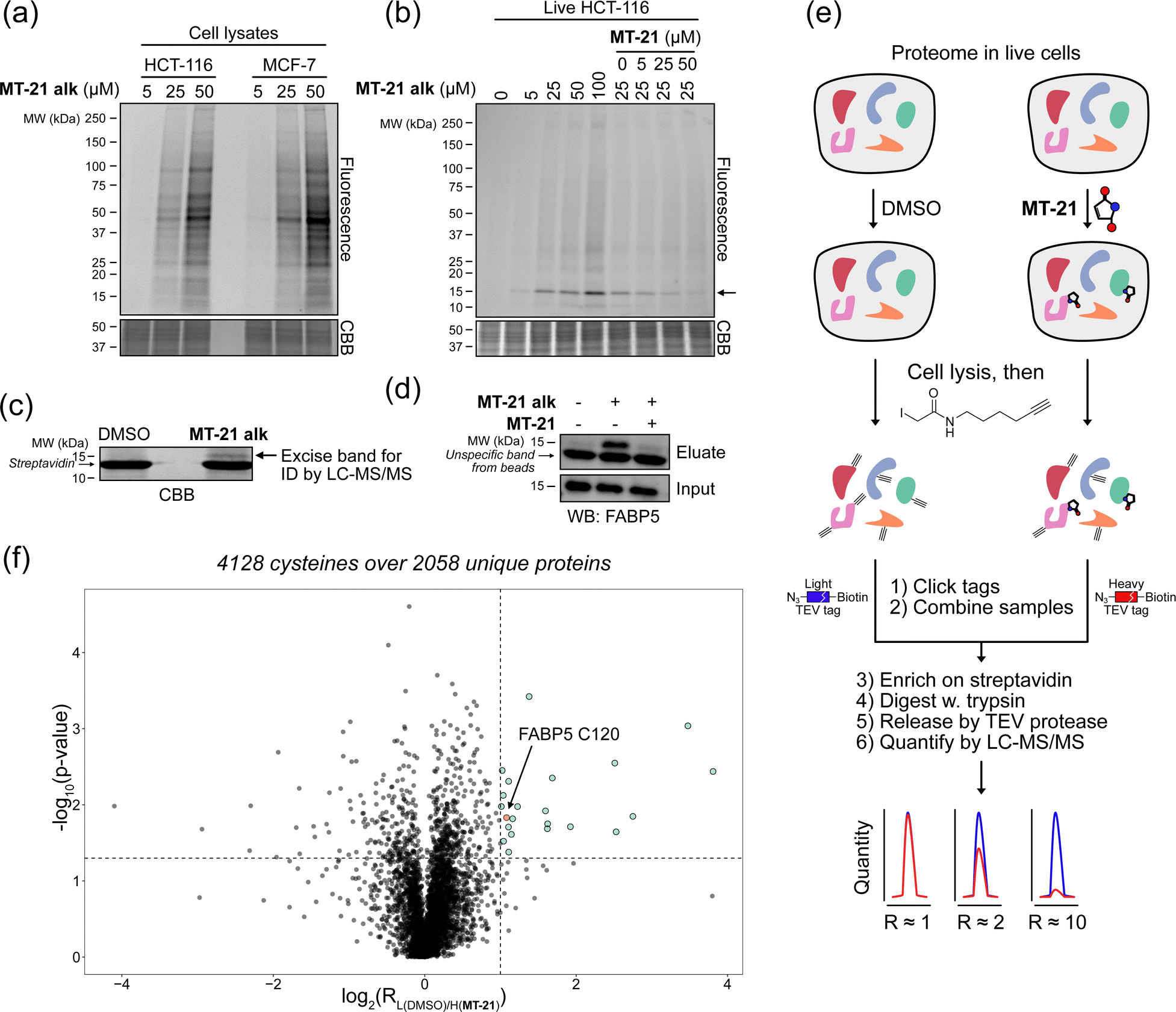 | ||
| Fig. 4 Proteomic reactivity of MT-21 alk. (a) Labelling with MT-21 alk in cell lysates from HCT-116 and MCF-7 cancer cell lines. The probe was incubated with lysate for 2 hours before CuAAC (click) reaction with TAMRA-N3 and SDS-PAGE followed by in gel fluorescence scanning. (b) Labelling with MT-21 alk in live HCT-116 cancer cells and competition with non-alkyne probe (MT-21). Cells were pre-treated with MT-21 or DMSO for 6 hours before addition of MT-21 alk for 12 hours, lysed and subjected to CuAAC with TAMRA-N3 and analyzed as in (a). (c) Identification of FABP5 as the 15 kDa protein by pulldown of covalently bound proteins. The 15 kDa band was excised from the gel and analyzed by LC-MS/MS. The strong, lower MW band is streptavidin from the beads (d) Confirmation of protein identification by pulldown followed by probing with an antibody against FABP5. The strong, lower MW band is an unspecific band from the beads, possibly released streptavidin. (e) Workflow for competitive cysteinome profiling by isoTOP-ABPP.14,15 (f) Profiling of the cysteinome reactivity of MT-21 by isoTOP-ABPP. Live cells were treated with 50 μM MT-21 or DMSO for 6 hours and then harvested and lysed. Free cysteines were labelled with iodoacetamide alkyne and clicked to isotopically differentiated TEV-protease-cleavable biotin-linked tags (heavy = MT-21, light = DMSO) and enriched for LC-MS/MS analysis. Significantly competed (log2(R) > 1, p-value < 0.05) peptides are shown as green dots. CBB = Coomassie Brilliant Blue. See ESI† for uncropped gels and for all experimental and analytical details. | ||
To provide a broader overview of binding proteins, we also performed an isoTOP-ABPP experiment14,15,52 (Fig. 4e) using iodoacetamide alkyne (IAyne) to probe for cysteine reactivity of MT-21. Live HCT-116 cells were with MT-21 treated for 6 hours before lysis and subsequent labelling of free cysteines with IAyne. Isotopically labelled biotin-azide tags bearing a TEV-protease cleavage site15 were then appended (light isotope-tag for DMSO, heavy for MT-21) via CuAAC. The samples were mixed and the competition was quantified using proteomics. In total we quantified 4128 cysteine-containing peptides distributed over 2058 proteins (raw data availble on OSF, see ESI†) from this experiment. We identified 22 significantly competed (light to heavy ratio > 2 and p-value < 0.05) peptides (Fig. 4f). For FABP5 we observed a light to heavy ratio of 2.1 (p-value = 0.015), indicating approximately 50% occupation at the conditions used in this experiment. The low occupation could be due to reversibility of the adduct. Nonetheless, we decided to look closer at the interaction between MT-21 and FABP5.
MT-21 covalently binds fatty acid binding protein 5 (FABP5) at cysteine 120
FABP5 (also named E-FABP, PA-FABP, K-FABP, C-FABP or mal1) is a lipid chaperone protein in the intracellular lipid-binding protein (iLBP) family, responsible for transporting fatty acids in cells.53 Besides its role in lipid transport, FAPB5 is important in lipid mediated signalling, where upon binding specific lipids, a conformational change takes place, which reveals a nuclear translocation signal. Upon nuclear translocation, FABP5 delivers its ligand and activates the nuclear receptors PPARγ54,55 or PPARβ/δ.56 FABP5 is not usually expressed in prostate cancer, but becomes highly expressed in metastatic prostate cancer.57 The protein has a total of six cysteines residues. The cysteine pair C120 and C127, located closely to the binding site of the carboxylate head group of fatty acids (Fig. S4, ESI†) shows interesting redox chemistry. In an early crystallographic study these two cysteines were found to form a disulphide bridge.58 However, in a more recent study, it was found that co-crystals with fatty acid ligands bound exclusively contained the free cysteines, while the apo-protein was found as a mixture.59 Furthermore, C127 oxidizes to the sulphenic acid upon treatment of RKO cells with H2O2.60 Contrary, C120 was found to lose reactivity upon treatment of A431 cells with EGF, which induces production of H2O2, indicative of oxidation.61 C120 is also reactive towards the broadly reactive metabolite 4-hydroxynonenal (4-HNE)62 possibly corroborating the finding in A431 cells. It is unknown if the redox chemistry between the C120–C127 pair can modulate ligand binding potency/selectivity, function as a regulatory element for FABP5-PPARβ/δ or PPARγ signalling or if the role is to scavenge reactive lipids as previously proposed.62To validate that MT-21 indeed binds in the established pocket of FABP5, we used the known FABP5/7 ligand SBFI-2663 to compete binding. Indeed, SBFI-26 was able to efficiently compete binding of MT-21 alk (Fig. 5a). The tryptic peptide identified in the mass spectrometry analysis contains cysteine-127 (C127) in addition to the putative target cysteine-120 (C120). To confirm modification of C120 by MT-21 alk, we expressed FLAG-tagged FABP5 with individual alanine-mutations of C120 and C127 as well as the double (C120A, C127A) mutant. We indeed observed covalent binding of MT-21 alk only when C120 was present (Fig. 5b) validating this as the binding site.
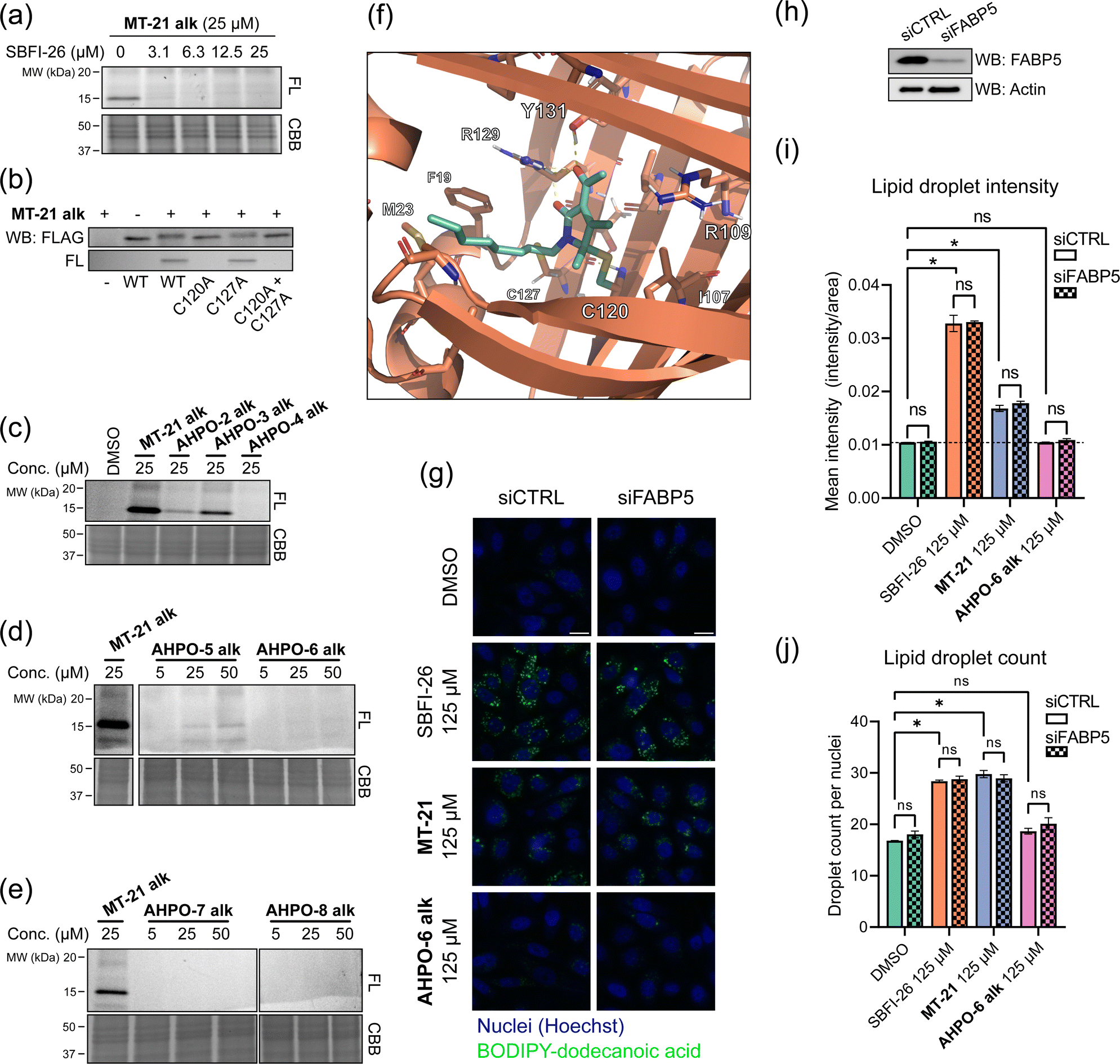 | ||
| Fig. 5 MT-21 covalently binds fatty acid-binding protein 5. (a) The known FABP5/7 ligand SBFI-26 competes the labelling of FABP5 by MT-21 alk. Labeling was visualized following CuAAC coupling of TAMRA-N3 and in-gel fluorescence scanning. (b) Validation of C120 as the target cysteine on FABP5. FLAG-tagged FABP5 was expressed in MCF-7 cells and treated with 100 μM MT-21 alk followed by TAMRA-N3 CuAAC and in-gel fluorescence. Only FABP5 variants bearing C120 could be covalently bound by MT-21 alk. (c–e) SAR of MT-21 alk on FABP5 in live HCT-116 cells with varying lipid-tail position (c), increased steric bulk at 4- and 5-position (d) and different electron withdrawing groups at position 3 (e). (f) Molecular dynamics modelling of FABP5 with covalently bound MT-21 through the hemiaminal carbon (g) lipid uptake assay with BODIPY-dodecanoic acid. PC-3 cells were transfected with control siRNA or siRNA targeting FABP5. After 48 hours the cells were seeded into a 96-well plate. The next day the cells were treated with SBFI-26, MT-21 and MT-21 5-Et alk for 2 hours in serum-free media followed by incubation with BODIPY-dodecanoic acid for 3 hours. The cells were then fixed, the nuclei were stained with DAPI and the cells were imaged. Scale bar = 20 μm. (h) Western blot confirms knock-down of FABP5. (i and j) Quantification of lipid droplets by image analysis. (i) Lipid droplet mean intensity. No significant difference is observed between control siRNA and siRNA targeting FABP5. Both MT-21 and SBFI-26 significantly increases the lipid droplet intensities compared to DMSO. See Fig. S10 (ESI†) for dose-response. Statistical analysis was performed with two-way ANOVA in GraphPad Prism 9. (j) Lipid droplet counts from, normalized to cell counts. Both MT-21 and SBFI-26 significantly increases the lipid droplet counts compared to DMSO. See Fig. S10 (ESI†) for dose-response. FL = Fluorescence, CBB = Coomassie Brilliant Blue. See ESI† for uncropped gels and membranes. | ||
MT-21 was originally synthesized along with a range of analogs with various N- and O-substitutions, all centered around the hemiaminal to mimic the natural product epolactaene.43 The analogs were initially evaluated as neurotrophic agents. Here, MT-21 scored particularly high. With this in mind, we next investigated how changes to the AHPO scaffold impact FABP5-binding. All compounds were evaluated in live HCT-116 cells (Fig. 5c–e, see also Fig. 2 for structures), and a selected subset of compounds also in a broader panel of cancer cell lines (Fig. S5, ESI†). Specifically, we envisioned that MT-21 would incorporate into or be associated with lipid membranes and that FABP5 extracts it from here followed by covalent conjugation in the bottom of the binding pocket, potentially assisted by a residue acting as a general acid catalyst. Such a mechanistic scenario could potentially explain the reduction in binding targets observed in live cells vs. cell lysate (Fig. 4a and b). Changing the relative position of the lipid/alkyne-tail and the AHPO-headgroup (AHPO-2 alk, AHPO-3 alk, AHPO-4 alk, see Fig. 2 for structures) would thus be expected to alter the reactivity and selectivity of protein binding, which would be reflected as changes to the observed labelling pattern. Towards this end, substitution on the ketone (AHPO-2 alk) gave a markedly loss of reactivity towards FABP5 and placing the lipid/alkyne-tail at the hemiaminal carbon C5 (AHPO-4 alk) completely abrogated labelling (Fig. 5c). In contrast, positioning of the lipid/alkyne-tail in position 4 (AHPO-3 alk) only afforded a small reduction in reactivity (Fig. 5c). We then tested the effect of minor changes to the steric bulk at C4 (and C5) by exchanging the methyl groups for ethyl groups, while maintaining the N-linked lipid chain present in MT-21 alk. Again, complete abrogation was observed upon increasing steric bulk at C5 (AHPO-6 alk) (Fig. 5d), but, curiously, we also observed significant loss of FABP5-reactivity with AHPO-5 alk (ethyl at C4). These results suggest that MT-21 has a near optimum geometry/reactivity for C120-modification and that only very minor tolerance exist for additional steric bulk. We finally tested whether the electron withdrawing group (EWG) at C3 had any impact on the reactivity: the methyl ketone of MT-21 alk was substituted for a methyl ester (AHPO-7 alk) or nitrile (AHPO-8 alk). In both cases, to our surprise, we saw complete loss of reactivity both in live cells (Fig. 5e), cell lysates and with methyl thioglycolate (Fig. S6, ESI†). This clearly indicates the privileged role of the C2 ketone group to unlock covalent reactivity of the AHPO-scaffold. Finally, to determine whether the stereocenter at C5 would have any effect on the labelling of FABP5, or other proteins, we incubated the previously purified enantiomers of MT-21 alk in live cells and cell lysates. However, we observed no differences in labelling intensity or kinetics of neither FABP5 nor the broad range of proteins bound in cell lysate (Fig. S2, ESI†) which suggest that racemization either occurs fast in these settings or concomitant with labelling.
Given the observed strong structural constraints for covalent binding between MT-21 and FABP5 in cells, we sought to obtain structural information on the conjugate. Specifically, we investigated the feasibility of co-crystallization of FABP5 and MT-21. We did this by incubating FABP5 with the probes MT-21 alk and AHPO-2 alk, the latter which does not bind FABP5 in whole cells (Fig. 5f), with recombinant FABP5 and assessed binding via in-gel fluorescence. Unlike the observation in cells, we observed similar labelling intensities with the two compounds (Fig. S7, ESI†) indicating unselective binding. We next heat-denatured FABP5, as previously described for CRABP2,29 but this also had no effect on labelling (Fig. S7, ESI†). These experiments again strongly indicate the importance of the cellular context for specific binding between MT-21 and FABP5, which discouraged co-crystallization studies, for which the complex would need to be formed in vitro with the recombinant protein. We instead conducted a molecular dynamics simulation (Fig. 5f and Fig. S8, ESI†). We modelled both the direct substitution on the hemiaminal carbon and the 1,6-addition followed by oxidation (mimicing the observed products with methyl thioglycolate) in both stereoisomeric outcomes, and performed explicit solvent all-atom molecular dynamics (MD) simulations. For the direct substitution product we observed instability of the “R”-product during the MD simulations (Fig. S8, ESI†). Contrary, the “S”-product was more stable. From the MD simulations we also observed stabilizing interactions between the dicarbonyl system of MT-21 and R129 and Y131, similar to what is observed in the crystal structure of linoleic acid bound to FABP5.59 The lipid tail of MT-21 exits the binding pocket in the same direction as linoleic acid, although it is not long enough to extend all the way to the solvent, through hydrophobic interactions with e.g. F19, M23 and V28.
Puzzled by the stark difference between the seemingly selective binding in whole cells and the promiscuous labelling pattern in cell lysates that we observed initially (Fig. 4a and b), we decided to take another look at the latter. Towards this end, pretreatment with iodoacetamide – to block reactive cysteines – only minimally diminished the engagement of MT-21 alk with proteins in cell lysates (Fig. S9, ESI†), indicating that these are either not cysteine targets (see Fig. 3c) or, alternatively, cysteine targets that are not efficiently engaged by iodoacetamide.20,27 To more directly probe the apparent loss in FABP5-reactivity in lysates, we first attempted pre-treatment with dithiotreitol (DTT) or TCEP, to reduce the potential C120–C127 disulphide bond but generally observed nearly complete loss of labelling (Fig. S9, ESI†). This could possibly be attributed by incompatibility of MT-21 alk with DTT/TCEP (Fig. S9, ESI†). We next labelled cell lysate from HCT-116 cells expressing FLAG-FABP5C127A, which cannot form a disulphide bond and is efficiently labeled by MT-21 alk in cells, but again observed no change in labelling pattern (Fig. S9, ESI†). An explanation for the observed lack of FABP5 reactivity could be occupation of FABP5 by an excess of liberated fatty acids present in lysate or, alternatively, that FABP5 needs interaction with a lipid bilayer to efficiently extract MT-21 alk.
MT-21 modulates cellular fatty acid-uptake in a FABP5-independent manner
We then investigated whether MT-21 caused any functional perturbation of lipid uptake/homeostasis and probed the involvement of the observed interaction with FABP5. We measured fatty acid uptake via a BODIPY-tagged dodecanoic acid analog in PC-3 cells and evaluated it by fluorescence microscopy in wild-type and FABP5 knock-down cells. We saw a strong increase in BODIPY intensity when treating with SBFI-26 and MT-21, while no increase was observed with AHPO-6 alk (Fig. 5g). No apparent visual difference between control siRNA and siRNA targeting FABP5 was, however, seen for SBFI-26 or MT-21. To obtain quantitative data, we analyzed the images in CellProfiler. On average, we quantified around 2700 cells for each condition, identifying around 22 lipid droplets per cell, totalling 54000 cells and 1.2 × 106 lipid droplets across the experiment. Confirming the qualitative, visual analysis, SBFI-26 and MT-21 significantly increased the mean intensity (Fig. 5i) and number of lipid droplets (Fig. 5j) in a dose-dependent manner (Fig. S10, ESI†) with no significant difference between control siRNA and siRNA targeting FABP5 (Fig. 5i and j) indicating that this phenotype is not dependent on FABP5. Previous reports indicate that FABP5 knockdown64 or inhibition by SBFI-2665 decrease fatty acid uptake, although others noted that fatty acid uptake is not perturbed by FABP5 knockdown.66 We cannot easily reconcile these differences in experimental observations, but our data indicates that SBFI-26 and MT-21 induce the observed phenotype (increase in fatty acid uptake) via a FABP5-independent mechanism, possibly by interaction with a related protein.
Conclusion
In summary, we show, through the use of a simplified model compound, MT-21, that the AHPO scaffold, which is present in various natural products, can be a biologically relevant electrophile. Specifically, our experiments demonstrate that the N-acyl-hemiaminal group can be activated under acidic conditions to – primarily – facilitate thiol reactivity. By separating the enantiomers of MT-21 alk we could investigate the activation mechanism in further detail, but curiously found no evidence of stereochemical instability under the acidic conditions used for thiol conjugation, indicating a direct substitution on the hemiaminal-carbon. Interestingly, no difference in proteome labelling was observed between the two enantiomers, possibly indicating that a fast racemization mechanism is operating in cells. When evaluated in biological settings, through the use of the alkyne-tagged analog, we initially observed a promiscuous labelling pattern in cell lysates. This, however, differed drastically from equivalent experiments conducted in live cells where a much more specific labelling pattern was observed with an apparent single protein preferentially labelled in gel-based experiments and only a few cysteines competed in the isoTOP-ABPP experiments. These experiments indicate that MT-21, possibly through membrane association, is exposed to a limited portion of the proteome in live cells. The single band observed in gel-based experiments was identified as the lipid chaperone FABP5 and we confirmed binding to cysteine 120, and not the neighbouring cysteine 127, through expression of FLAG-tagged mutants of FABP5. This cysteine pair, which is known to undergo reversible disulfide formation, may have a direct role in the cellular functions of FABP5 and our results suggest that MT-21 might be useful as a molecular probe to study this potential regulatory element. Considering the role of FABP5 in transporting arachidonic acid,67 a precurser of the endocannabinoid neurotransmitter anandamide, and retinoic acid,68,69 a vitamin A1 metabolite involved in neurogenesis,70,71 the observed neurotrophic effects of the epolactene derivatives,43 including MT-21, might be explained by their modulation of this protein and this represent one interesting future research direction. We cannot refute that MT-21 engages ANT-1 as originally reported by Osada and co-workers.47 However, our studies clearly show that the compound also engages other targets, some covalently, and we therefore discourage the use of MT-21 as a selective ANT-1 modulator.47Data availability
Raw data, including gel images and western blot membranes, and scripts for data analysis are available at OSF (DOI: 10.17605/OSF.IO/U5S4C). Raw images for fluorescence microscopy is available at Zenodo (see ESI†). Raw proteomics data and MaxQuant parameter file for isoTOP-ABPP is available at OSF (DOI: 10.17605/OSF.IO/U5S4C).Author contributions
E. B. S. and T. B. P. designed experiments. E. B. S., K. H. J., and B. K. H. performed biological experiments. RNO performed organic synthesis. M. N., J. P. and S. M. H. performed proteomics data acquisition and data analysis. C. K. performed cloning and protein expression under supervision of T. T. Y. W. and K. L. L. performed molecular modelling and dynamics simulations. E. B. S. prepared figures. E. B. S. and T. B. P. wrote the manuscript. All authors proof-read the manuscript and relevant parts of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
Thanks to Prof. Daniel Otzen at the Interdisciplinary Nanoscience Center, Aarhus University, Denmark for use of circular dichorism spectrometer. Thanks to Prof. Karl Anker Jørgensen at the Department of Chemistry, Aarhus University, Denmark for use UPCC for chiral chromatography. TBP acknowleges support from the Carlsberg Foundation (grant CF17-800) and Novo Nordisk Foundation (grant NNF19OC0054782). YW and KLL were supported by the BRAINSTRUC initiative from the Lundbeck Foundation (R155-2015-2666). YW and KLL acknowledge access to computational resources from the Danish National Supercomputer for Life Sciences (Computerome) and the ROBUST Resource for Biomolecular Simulations (supported by the Novo Nordisk Foundation; NNF18OC0032608).References
- J. Singh, R. C. Petter, T. A. Baillie and A. Whitty, The Resurgence of Covalent Drugs, Nat. Rev. Drug Discov., 2011, 10, 307–317, DOI:10.1038/nrd3410.
- F. Sutanto, M. Konstantinidou and A. Dömling, Covalent Inhibitors: A Rational Approach to Drug Discovery, RSC Med. Chem., 2020, 11, 876–884, 10.1039/D0MD00154F.
- J. N. Spradlin, E. Zhang and D. K. Nomura, Reimagining Druggability Using Chemoproteomic Platforms, Acc. Chem. Res., 2021, 54, 1801–1813, DOI:10.1021/acs.accounts.1c00065.
- T. Tamura and I. Hamachi, Chemistry for Covalent Modification of Endogenous/Native Proteins: From Test Tubes to Complex Biological Systems, J. Am. Chem. Soc., 2019, 141, 2782–2799, DOI:10.1021/jacs.8b11747.
- L. Xu, S. L. Kuan and T. Weil, Contemporary Approaches for Site-Selective Dual Functionalization of Proteins, Angew. Chem., Int. Ed., 2021, 60, 13757–13777, DOI:10.1002/anie.202012034.
- E. A. Hoyt, P. M. S. D. Cal, B. L. Oliveira and G. J. L. Bernardes, Contemporary Approaches to Site-Selective Protein Modification, Nat. Rev. Chem., 2019, 3, 147–171, DOI:10.1038/s41570-019-0079-1.
- J. N. Spradlin, X. Hu, C. C. Ward, S. M. Brittain, M. D. Jones, L. Ou, M. To, A. Proudfoot, E. Ornelas, M. Woldegiorgis, J. A. Olzmann, D. E. Bussiere, J. R. Thomas, J. A. Tallarico, J. M. McKenna, M. Schirle, T. J. Maimone and D. K. Nomura, Harnessing the Anti-Cancer Natural Product Nimbolide for Targeted Protein Degradation, Nat. Chem. Biol., 2019, 15, 747–755, DOI:10.1038/s41589-019-0304-8.
- R. Gabizon and N. London, The Rise of Covalent Proteolysis Targeting Chimeras, Curr. Opin. Chem. Biol., 2021, 62, 24–33, DOI:10.1016/j.cbpa.2020.12.003.
- C. C. Ward, J. I. Kleinman, S. M. Brittain, P. S. Lee, C. Y. S. Chung, K. Kim, Y. Petri, J. R. Thomas, J. A. Tallarico, J. M. McKenna, M. Schirle and D. K. Nomura, Covalent Ligand Screening Uncovers a RNF4 E3 Ligase Recruiter for Targeted Protein Degradation Applications, ACS Chem. Biol., 2019, 14, 2430–2440, DOI:10.1021/acschembio.8b01083.
- N. J. Henning, L. Boike, J. N. Spradlin, C. C. Ward, G. Liu, E. Zhang, B. P. Belcher, S. M. Brittain, M. J. Hesse, D. Dovala, L. M. McGregor, R. Valdez Misiolek, L. W. Plasschaert, D. J. Rowlands, F. Wang, A. O. Frank, D. Fuller, A. R. Estes, K. L. Randal, A. Panidapu, J. M. McKenna, J. A. Tallarico, M. Schirle and D. K. Nomura, Deubiquitinase-Targeting Chimeras for Targeted Protein Stabilization, Nat. Chem. Biol., 2022, 18, 412–421, DOI:10.1038/s41589-022-00971-2.
- E. Resnick, A. Bradley, J. Gan, A. Douangamath, T. Krojer, R. Sethi, P. P. Geurink, A. Aimon, G. Amitai, D. Bellini, J. Bennett, M. Fairhead, O. Fedorov, R. Gabizon, J. Gan, J. Guo, A. Plotnikov, N. Reznik, G. F. Ruda, L. Díaz-Sáez, V. M. Straub, T. Szommer, S. Velupillai, D. Zaidman, Y. Zhang, A. R. Coker, C. G. Dowson, H. M. Barr, C. Wang, K. V. M. Huber, P. E. Brennan, H. Ovaa, F. von Delft and N. London, Rapid Covalent-Probe Discovery by Electrophile-Fragment Screening, J. Am. Chem. Soc., 2019, 141, 8951–8968, DOI:10.1021/jacs.9b02822.
- M. B. Cordon, K. M. Jacobsen, C. S. Nielsen, P. Hjerrild and T. B. Poulsen, Forward Chemical Genetic Screen for Oxygen-Dependent Cytotoxins Uncovers New Covalent Fragments That Target GPX4, ChemBioChem, 2022, 23, cbic.202100253, DOI:10.1002/cbic.202100253.
- W. Lu, M. Kostic, T. Zhang, J. Che, M. P. Patricelli, L. H. Jones, E. T. Chouchani and N. S. Gray, Fragment-Based Covalent Ligand Discovery, RSC Chem. Biol., 2021, 2, 354–367, 10.1039/D0CB00222D.
- K. M. Backus, B. E. Correia, K. M. Lum, S. Forli, B. D. Horning, G. E. González-Páez, S. Chatterjee, B. R. Lanning, J. R. Teijaro, A. J. Olson, D. W. Wolan and B. F. Cravatt, Proteome-Wide Covalent Ligand Discovery in Native Biological Systems, Nature, 2016, 534, 570–574, DOI:10.1038/nature18002.
- E. Weerapana, C. Wang, G. M. Simon, F. Richter, S. Khare, M. B. D. Dillon, D. A. Bachovchin, K. Mowen, D. Baker and B. F. Cravatt, Quantitative Reactivity Profiling Predicts Functional Cysteines in Proteomes, Nature, 2010, 468, 790–795, DOI:10.1038/nature09472.
- P. R. A. Zanon, L. Lewald and S. M. Hacker, Isotopically Labeled Desthiobiotin Azide (IsoDTB) Tags Enable Global Profiling of the Bacterial Cysteinome, Angew. Chem., Int. Ed., 2020, 59, 2829–2836, DOI:10.1002/anie.201912075.
- M. Gehringer and S. A. Laufer, Emerging and Re-Emerging Warheads for Targeted Covalent Inhibitors: Applications in Medicinal Chemistry and Chemical Biology, J. Med. Chem., 2019, 62, 5673–5724, DOI:10.1021/acs.jmedchem.8b01153.
- T. Zhang, J. M. Hatcher, M. Teng, N. S. Gray and M. Kostic, Recent Advances in Selective and Irreversible Covalent Ligand Development and Validation, Cell Chem. Biol., 2019, 26, 1486–1500, DOI:10.1016/j.chembiol.2019.09.012.
- D. A. Shannon and E. Weerapana, Covalent Protein Modification: The Current Landscape of Residue-Specific Electrophiles, Curr. Opin. Chem. Biol., 2015, 24, 18–26, DOI:10.1016/j.cbpa.2014.10.021.
- P. R. A. Zanon, F. Yu, P. Z. Musacchio, L. Lewald, M. Zollo, K. Krauskopf, D. Mrdović, P. Raunft, T. E. Maher, M. Cigler, C. J. Chang, K. Lang, F. D. Toste, A. I. Nesvizhskii and S. M. Hacker, Profiling the Proteome-Wide Selectivity of Diverse Electrophiles, ChemRxiv, 2021, 1, 1–10, DOI:10.26434/chemrxiv-2021-w7rss-v2.
- M. R. Wilson, Y. Jiang, P. W. Villalta, A. Stornetta, P. D. Boudreau, A. Carrá, C. A. Brennan, E. Chun, L. Ngo, L. D. Samson, B. P. Engelward, W. S. Garrett, S. Balbo and E. P. Balskus, The Human Gut Bacterial Genotoxin Colibactin Alkylates DNA, Science, 2019, 363, eaar7785, DOI:10.1126/science.aar7785.
- M. Xue, C. S. Kim, A. R. Healy, K. M. Wernke, Z. Wang, M. C. Frischling, E. E. Shine, W. Wang, S. B. Herzon and J. M. Crawford, Structure Elucidation of Colibactin and Its DNA Cross-Links, Science, 2019, 365, eaax2685, DOI:10.1126/science.aax2685.
- Y. Isobe, M. Okumura, L. M. McGregor, S. M. Brittain, M. D. Jones, X. Liang, R. White, W. Forrester, J. M. McKenna, J. A. Tallarico, M. Schirle, T. J. Maimone and D. K. Nomura, Manumycin Polyketides Act as Molecular Glues between UBR7 and P53, Nat. Chem. Biol., 2020, 16, 1189–1198, DOI:10.1038/s41589-020-0557-2.
- G. J. Wørmer, N. L. Villadsen, P. Nørby and T. B. Poulsen, Concise Asymmetric Syntheses of Streptazone A and Abikoviromycin, Angew. Chem., Int. Ed., 2021, 60, 10521–10525, DOI:10.1002/anie.202101439.
- H. Liu, R. N. Ottosen, K. M. Jennet, E. B. Svenningsen, T. F. Kristensen, M. Biltoft, M. R. Jakobsen and T. B. Poulsen, Macrodiolide Diversification Reveals Broad Immunosuppressive Activity That Impairs the CGAS-STING Pathway, Angew. Chem., Int. Ed., 2021, 60, 18734–18741, DOI:10.1002/anie.202105793.
- K. M. Jacobsen, N. L. Villadsen, T. Tørring, C. B. Nielsen, T. Salomón, M. M. Nielsen, M. Tsakos, C. Sibbersen, C. Scavenius, R. Nielsen, E. I. Christensen, P. F. Guerra, P. Bross, J. S. Pedersen, J. J. Enghild, M. Johannsen, J. Frøkiær, J. Overgaard, M. R. Horsman, M. Busk and T. B. Poulsen, APD-Containing Cyclolipodepsipeptides Target Mitochondrial Function in Hypoxic Cancer Cells, Cell Chem. Biol., 2018, 25, 1337–1349.e12, DOI:10.1016/j.chembiol.2018.07.010.
- D. Abegg, M. Tomanik, N. Qiu, D. Pechalrieu, A. Shuster, B. Commare, A. Togni, S. B. Herzon and A. Adibekian, Chemoproteomic Profiling by Cysteine Fluoroalkylation Reveals Myrocin G as an Inhibitor of the Nonhomologous End Joining DNA Repair Pathway, J. Am. Chem. Soc., 2021, 143, 20332–20342, DOI:10.1021/jacs.1c09724.
- J. Dong, L. Krasnova, M. G. Finn and K. B. Sharpless, Sulfur(VI) Fluoride Exchange (SuFEx): Another Good Reaction for Click Chemistry, Angew. Chem., Int. Ed., 2014, 53, 9430–9448, DOI:10.1002/anie.201309399.
- W. Chen, J. Dong, L. Plate, D. E. Mortenson, G. J. Brighty, S. Li, Y. Liu, A. Galmozzi, P. S. Lee, J. J. Hulce, B. F. Cravatt, E. Saez, E. T. Powers, I. A. Wilson, K. B. Sharpless and J. W. Kelly, Arylfluorosulfates Inactivate Intracellular Lipid Binding Protein(s) through Chemoselective SuFEx Reaction with a Binding Site Tyr Residue, J. Am. Chem. Soc., 2016, 138, 7353–7364, DOI:10.1021/jacs.6b02960.
- J. K. Eaton, L. Furst, R. A. Ruberto, D. Moosmayer, A. Hilpmann, M. J. Ryan, K. Zimmermann, L. L. Cai, M. Niehues, V. Badock, A. Kramm, S. Chen, R. C. Hillig, P. A. Clemons, S. Gradl, C. Montagnon, K. E. Lazarski, S. Christian, B. Bajrami, R. Neuhaus, A. L. Eheim, V. S. Viswanathan and S. L. Schreiber, Selective Covalent Targeting of GPX4 Using Masked Nitrile-Oxide Electrophiles, Nat. Chem. Biol., 2020, 16, 497–506, DOI:10.1038/s41589-020-0501-5.
- B. K. Hansen, C. J. Loveridge, S. Thyssen, G. J. Wørmer, A. D. Nielsen, J. Palmfeldt, M. Johannsen and T. B. Poulsen, STEFs: Activated Vinylogous Protein-Reactive Electrophiles, Angew. Chem., Int. Ed., 2019, 58, 3533–3537, DOI:10.1002/anie.201814073.
- G. J. Wørmer, B. K. Hansen, J. Palmfeldt and T. B. Poulsen, A Cyclopropene Electrophile That Targets Glutathione S-Transferase Omega-1 in Cells, Angew. Chem., Int. Ed., 2019, 58, 11918–11922, DOI:10.1002/anie.201907520.
- Y. Qian, M. Schürmann, P. Janning, C. Hedberg and H. Waldmann, Activity-Based Proteome Profiling Probes Based on Woodward's Reagent K with Distinct Target Selectivity, Angew. Chem., Int. Ed., 2016, 55, 7766–7771, DOI:10.1002/anie.201602666.
- B. Nay, N. Riache and L. Evanno, Chemistry and Biology of Non-Tetramic γ-Hydroxy-γ-Lactams and γ-Alkylidene-γ-Lactams from Natural Sources, Nat. Prod. Rep., 2009, 26, 1044–1062, 10.1039/B903905H.
- R. Shiraki, A. Sumino, K. Tadano and S. Ogawa, Total Synthesis of PI-091, Tetrahedron Lett., 1995, 36, 5551–5554, DOI:10.1016/0040-4039(95)01049-N.
- R. Shiraki, A. Sumino, K. Tadano and S. Ogawa, Total Synthesis of Natural PI-091, a New Platelet Aggregation Inhibitor of Microbial Origin, J. Org. Chem., 1996, 61, 2845–2852, DOI:10.1021/jo951897z.
- S. B. Singh, M. A. Goetz, E. T. Jones, G. F. Bills, R. A. Giacobbe, L. Herranz, S. Stevens-Miles and D. L. Williams, Oteromycin: A Novel Antagonist of Endothelin Receptor, J. Org. Chem., 1995, 60, 7040–7042, DOI:10.1021/jo00126a071.
- H. He, H. Y. Yang, R. Bigelis, E. H. Solum, M. Greenstein and G. T. Carter, Pyrrocidines A and B, New Antibiotics Produced by a Filamentous Fungus, Tetrahedron Lett., 2002, 43, 1633–1636, DOI:10.1016/S0040-4039(02)00099-0.
- S. Uesugi, N. Fujisawa, J. Yoshida, M. Watanabe, S. Dan, T. Yamori, Y. Shiono and K. I. Kimura, Pyrrocidine A, a Metabolite of Endophytic Fungi, Has a Potent Apoptosis-Inducing Activity against HL60 Cells through Caspase Activation via the Michael Addition, J. Antibiot., 2016, 69, 133–140, DOI:10.1038/ja.2015.103.
- D. A. Burnett, J. K. Choi, D. J. Hart and Y. M. Tsai, Pyrrolizidinone and Indolizidinone Synthesis: Generation and Intramolecular Addition of Alpha-Acylamino Radicals to Olefins and Allenes, J. Am. Chem. Soc., 1984, 106, 8201–8209, DOI:10.1021/ja00338a033.
- P. Pigeon and B. Decroix, Tetracyclic Systems: Synthesis of Isoindolo[1,2-b]Thieno-[2,3(3,2 or 3,4)-e][1,3]Thiazocines and Isoindolo[2,1-a]Thieno-[2,3(3,2 or 3,4)-f ][1,4] and [1,5]Diazocines, J. Heterocycl. Chem., 1997, 34, 375–380, DOI:10.1002/jhet.5570340203.
- R. Adhikari, D. A. Jones, A. J. Liepa and R. H. Nearn, Synthesis of N-Substituted γ-Methylene γ-Lactams, Aust. J. Chem., 2005, 58, 882, DOI:10.1071/CH05286.
- H. Kakeya, C. Onozawa, M. Sato, K. Arai and H. Osada, Neuritogenic Effect of Epolactaene Derivatives on Human Neuroblastoma Cells Which Lack High-Affinity Nerve Growth Factor Receptors, J. Med. Chem., 1997, 40, 391–394, DOI:10.1021/jm960719a.
- H. Kakeya, I. Takahasi, G. Okada, K. Isono and H. Osada, Epolactaene, a Novel Neuritogenic Compound in Human Neuroblastoma Cells, Produced by a Marine Fungus, J. Antibiot., 1995, 48, 733–735, DOI:10.7164/antibiotics.48.733.
- M. Watabe, H. Kakeya and H. Osada, Requirement of Protein Kinase (Krs/MST) Activation for MT-21-Induced Apoptosis, Oncogene, 1999, 18, 5211–5220, DOI:10.1038/sj.onc.1202901.
- M. Watabe, K. Machida and H. Osada, MT-21 Is a Synthetic Apoptosis Inducer That Directly Induces Cytochrome c Release from Mitochondria, Cancer Res., 2000, 60, 5214–5222 CAS.
- K. Machida, Y. Hayashi and H. Osada, A Novel Adenine Nucleotide Translocase Inhibitor, MT-21, Induces Cytochrome c Release by a Mitochondrial Permeability Transition-Independent Mechanism, J. Biol. Chem., 2002, 277, 31243–31248, DOI:10.1074/jbc.M204564200.
- C. A. Brown and A. Yamashita, Saline Hydrides and Superbases in Organic Reactions. IX. Acetylene Zipper. Exceptionally Facile Contrathermodynamic Multipositional Isomeriazation of Alkynes with Potassium 3-Aminopropylamide, J. Am. Chem. Soc., 1975, 97, 891–892, DOI:10.1021/ja00837a034.
- D. M. Birney, X. Xu, S. Ham and X. Huang, Chemoselectivity in the Reactions of Acetylketene and Acetimidoylketene: Confirmation of Theoretical Predictions, J. Org. Chem., 1997, 62, 7114–7120, DOI:10.1021/jo971083d.
- A. L. García-Cabeza, R. Marín-Barrios, R. Azarken, F. J. Moreno-Dorado, M. J. Ortega, H. Vidal, J. M. Gatica, G. M. Massanet and F. M. Guerra, DoE (Design of Experiments) Assisted Allylic Hydroxylation of Enones Catalysed by a Copper-Aluminium Mixed Oxide, European J. Org. Chem., 2013, 3, 8307–8314, DOI:10.1002/ejoc.201301145.
- D. Kalaitzakis, A. Kouridaki, D. Noutsias, T. Montagnon and G. Vassilikogiannakis, Methylene Blue as a Photosensitizer and Redox Agent: Synthesis of 5-Hydroxy-1H-Pyrrol-2(5H)-Ones from Furans, Angew. Chem., Int. Ed., 2015, 54, 6283–6287, DOI:10.1002/anie.201500744.
- C. Wang, E. Weerapana, M. M. Blewett and B. F. Cravatt, A Chemoproteomic Platform to Quantitatively Map Targets of Lipid-Derived Electrophiles, Nat. Methods, 2014, 11, 79–85, DOI:10.1038/nmeth.2759.
- R. L. Smathers and D. R. Petersen, The Human Fatty Acid-Binding Protein Family: Evolutionary Divergences and Functions, Hum. Genomics, 2011, 5, 170, DOI:10.1186/1479-7364-5-3-170.
- Z. Bao, M. I. Malki, S. S. Forootan, J. Adamson, F. S. Forootan, D. Chen, C. S. Foster, P. S. Rudland and Y. Ke, A Novel Cutaneous Fatty Acid-Binding Protein-Related Signaling Pathway Leading to Malignant Progression in Prostate Cancer Cells, Genes Cancer, 2013, 4, 297–314, DOI:10.1177/1947601913499155.
- F. S. Forootan, S. S. Forootan, X. Gou, J. Yang, B. Liu, D. Chen, M. S. Al Fayi, W. Al-Jameel, P. S. Rudland, S. A. Hussain and Y. Ke, Fatty Acid Activated PPARf Promotes Tumorigenicity of Prostate Cancer Cells by up Regulating VEGF via PPAR Responsive Elements of the Promoter, Oncotarget, 2016, 7, 9322–9339, DOI:10.18632/oncotarget.6975.
- J. L. Napoli, Cellular Retinoid Binding-Proteins, CRBP, CRABP, FABP5: Effects on Retinoid Metabolism, Function and Related Diseases, Pharmacol. Ther., 2017, 173, 19–33, DOI:10.1016/j.pharmthera.2017.01.004.
- G. Carbonetti, T. Wilpshaar, J. Kroonen, K. Studholme, C. Converso, S. D’Oelsnitz and M. Kaczocha, FABP5 Coordinates Lipid Signaling That Promotes Prostate Cancer Metastasis, Sci. Rep., 2019, 9, 18944, DOI:10.1038/s41598-019-55418-x.
- C. Hohoff, T. Börchers, B. Rüstow, F. Spener and H. Van Tilbeurgh, Expression, Purification, and Crystal Structure Determination of Recombinant Human Epidermal-Type Fatty Acid Binding Protein, Biochemistry, 1999, 38, 12229–12239, DOI:10.1021/bi990305u.
- E. H. Armstrong, D. Goswami, P. R. Griffin, N. Noy and E. A. Ortlund, Structural Basis for Ligand Regulation of the Fatty Acid-Binding Protein 5, Peroxisome Proliferator-Activated Receptor β/δ (FABP5-PPARβ/δ) Signaling Pathway, J. Biol. Chem., 2014, 289, 14941–14954, DOI:10.1074/jbc.M113.514646.
- J. Yang, V. Gupta, K. S. Carroll and D. C. Liebler, Site-Specific Mapping and Quantification of Protein S-Sulphenylation in Cells, Nat. Commun., 2014, 5, 4776, DOI:10.1038/ncomms5776.
- M. Abo and E. Weerapana, A Caged Electrophilic Probe for Global Analysis of Cysteine Reactivity in Living Cells, J. Am. Chem. Soc., 2015, 137, 7087–7090, DOI:10.1021/jacs.5b04350.
- A. Bennaars-Eiden, L. Higgins, A. V. Hertzel, R. J. Kapphahn, D. A. Ferrington and D. A. Bernlohr, Covalent Modification of Epithelial Fatty Acid-Binding Protein by 4-Hydroxynonenal in Vitro and in Vivo, J. Biol. Chem., 2002, 277, 50693–50702, DOI:10.1074/jbc.M209493200.
- M. Kaczocha, M. J. Rebecchi, B. P. Ralph, Y.-H. G. Teng, W. T. Berger, W. Galbavy, M. W. Elmes, S. T. Glaser, L. Wang, R. C. Rizzo, D. G. Deutsch and I. Ojima, Inhibition of Fatty Acid Binding Proteins Elevates Brain Anandamide Levels and Produces Analgesia, PLoS One, 2014, 9, e94200, DOI:10.1371/journal.pone.0094200.
- T. Wu, J. Tian, R. G. Cutler, R. S. Telljohann, D. A. Bernlohr, M. P. Mattson and J. T. Handa, Knockdown of FABP5 MRNA Decreases Cellular Cholesterol Levels and Results in Decreased ApoB100 Secretion and Triglyceride Accumulation in ARPE-19 Cells, Lab. Investig., 2010, 90, 906–914, DOI:10.1038/labinvest.2009.33.
- W. Al-Jameel, X. Gou, S. S. Forootan, M. S. Al Fayi, P. S. Rudland, F. S. Forootan, J. Zhang, P. A. Cornford, S. A. Hussain and Y. Ke, Inhibitor SBFI26 Suppresses the Malignant Progression of Castration-Resistant PC3-M Cells by Competitively Binding to Oncogenic FABP5, Oncotarget, 2017, 8, 31041–31056, DOI:10.18632/oncotarget.16055.
- S. Senga, N. Kobayashi, K. Kawaguchi, A. Ando and H. Fujii, Fatty Acid-Binding Protein 5 (FABP5) Promotes Lipolysis of Lipid Droplets, de Novo Fatty Acid (FA) Synthesis and Activation of Nuclear Factor-Kappa B (NF-KB) Signaling in Cancer Cells, Biochim. Biophys. Acta, Mol. Cell Biol. Lipids, 1863, 2018, 1057–1067, DOI:10.1016/j.bbalip.2018.06.010.
- J.-W. Liu, F. G. Almaguel, L. Bu, D. D. De Leon and M. De Leon, Expression of E-FABP in PC12 Cells Increases Neurite Extension during Differentiation: Involvement of n-3 and n-6 Fatty Acids, J. Neurochem, 2008, 106, 2015–2029, DOI:10.1111/j.1471-4159.2008.05507.x.
- T. T. Schug, D. C. Berry, N. S. Shaw, S. N. Travis and N. Noy, Opposing Effects of Retinoic Acid on Cell Growth Result from Alternate Activation of Two Different Nuclear Receptors, Cell, 2007, 129, 723–733, DOI:10.1016/j.cell.2007.02.050.
- N.-S. Tan, N. S. Shaw, N. Vinckenbosch, P. Liu, R. Yasmin, B. Desvergne, W. Wahli and N. Noy, Selective Cooperation between Fatty Acid Binding Proteins and Peroxisome Proliferator-Activated Receptors in Regulating Transcription, Mol. Cell. Biol., 2002, 22, 5114–5127, DOI:10.1128/MCB.22.14.5114-5127.2002.
- S. Yu, L. Levi, R. Siegel and N. Noy, Retinoic Acid Induces Neurogenesis by Activating Both Retinoic Acid Receptors (RARs) and Peroxisome Proliferator-Activated Receptor β/δ (PPARβ/δ, J. Biol. Chem., 2012, 287, 42195–42205, DOI:10.1074/jbc.M112.410381.
- R. A. Merrill, J. M. Ahrens, M. E. Kaiser, K. S. Federhart, V. Y. Poon and M. Clagett-Dame, All-Trans Retinoic Acid-Responsive Genes Identified in the Human SH-SY5Y Neuroblastoma Cell Line and Their Regulated Expression in the Nervous System of Early Embryos, Biol. Chem., 2004, 385, 605–614, DOI:10.1515/BC.2004.075.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2cb00161f |
| ‡ Current address: College of Life Sciences, Zhejiang University, Hangzhou 310027, China. |
| This journal is © The Royal Society of Chemistry 2022 |