
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCereblon covalent modulation through structure-based design of histidine targeting chemical probes†
Justin T.
Cruite
*ab,
Geoffrey P.
Dann
*ab,
Jianwei
Che
ab,
Katherine A.
Donovan
bc,
Silas
Ferrao
a,
Scott B.
Ficarro
de,
Eric S.
Fischer
 abc,
Nathanael S.
Gray
f,
Fidel
Huerta
a,
Nikki R.
Kong
ab,
Hu
Liu
ab,
Jarrod A.
Marto
de,
Rebecca J.
Metivier
c,
Radosław P.
Nowak
ab,
Breanna L.
Zerfas
ab and
Lyn H.
Jones
*ab
abc,
Nathanael S.
Gray
f,
Fidel
Huerta
a,
Nikki R.
Kong
ab,
Hu
Liu
ab,
Jarrod A.
Marto
de,
Rebecca J.
Metivier
c,
Radosław P.
Nowak
ab,
Breanna L.
Zerfas
ab and
Lyn H.
Jones
*ab
aCenter for Protein Degradation, Dana-Farber Cancer Institute, Boston, MA, USA. E-mail: lyn_jones@dfci.harvard.edu
bDepartment of Biological Chemistry and Molecular Pharmacology, Harvard Medical School, Boston, MA, USA
cDepartment of Cancer Biology, Dana-Farber Cancer Institute, Boston, MA, USA
dDepartment of Cancer Biology, Department of Oncologic Pathology, and Blais Proteomics Center, Dana-Farber Cancer Institute, Boston, MA, USA
eDepartment of Pathology, Brigham and Women's Hospital, Harvard Medical School, Boston, MA 02115, USA
fDepartment of Chemical and Systems Biology, ChEM-H, Stanford Cancer Institute, School of Medicine, Stanford University, Stanford, CA, USA
First published on 8th July 2022
Abstract
Electrophilic biocompatible warheads, particularly cysteine-reactive acrylamides, have enabled the development of covalent inhibitor drugs and chemical biology probes, but cysteine is rarely present in protein binding sites. Therefore, expansion of the list of targetable amino acid residues is required to augment the synthetic bology toolkit of site-selective protein modifications. This work describes the first rational targeting of a specific histidine residue in a protein binding site using sulfonyl exchange chemistry. Structure-based drug design was used to incorporate sulfonyl fluoride and triazole reactive groups into the isoindolinone thalidomide congener EM12 to yield potent covalent inhibitors of the cereblon E3 ubiquitin ligase complex through engagement of His353. Conversely, the fluorosulfate derivative EM12-FS labels His353, but degrades a novel neosubstrate, the protein N-terminal glutamine amidohydrolase NTAQ1, which is involved in the N-end rule pathway and DNA damage response. Targeted protein degradation using cereblon ligands has become an important new drug discovery modality and the chemical probes and covalent labeling strategy described here will broadly impact this exciting area of therapeutic research.
Introduction
Targeted covalent inhibitors of proteins often possess enhanced pharmacological effects driven by improvements in potency, selectivity and pharmacodynamic duration compared to reversible binders.1–3 The rational targeting of cysteine in protein binding sites has led to the successful development of numerous covalent drugs and chemical probes.4,5 However, cysteine may not present itself as a targetable residue due to its rarity in protein binding sites.6,7 Additionally, the high intrinsic nucleophilicity of the cysteine thiol functionality creates challenges for the development of selective inhibitors. As a result, alternative approaches are required to advance small molecule modulators that engage residues beyond cysteine.8,9 Sulfur(VI)-fluoride exchange (SuFEx) chemistry has shown considerable promise as a synthetic clickable hub10 and a chemical biology platform with impactful applications in drug discovery.11,12 In particular, sulfonyl fluoride and fluorosulfate electrophilic warheads incorporated into small molecule ligands have been shown to site-selectively modify multiple residues on diverse proteins in cells, including tyrosine, lysine and serine.11,12 The targeting of histidine is relatively understudied,1,9 despite its preponderance in protein active sites, often acting as an acid–base catalyst due to its amphoteric properties, or as a catalytic nucleophile in RNA/DNA-binding proteins.13,14 Histidine is also frequently proximal to drugs and drug-like molecules in protein binding sites.15 The covalent ATP mimic 5′-fluorosulfonylbenzoyl 5′-adenosine (FSBA), that preferentially labels tyrosine and lysine, was shown previously to fortuitously engage a histidine residue in the mitochondrial F1-ATPase enzyme.16 A functionally important histidine that interacts with ATP in the binding pocket of Salmonella typhimurium 5-phosphoribosyl-α-1-pyrophosphate (PRPP) synthetase was also found to be labelled by FSBA.17 These serendipitous discoveries demonstrate the potential for sulfonyl fluorides to modify histidine side chains,18 but the rational targeting of specific endogenous histidine residues in proteins has not been reported. A variation of the SuFEx strategy called sulfur-triazole exchange (SuTEx) chemistry was recently reported where the fluorine is replaced by triazole heteroaromatic rings that enable further tuning of the leaving group to modulate reactivity and specificity.19–21 Promiscuous SuTEx probes were shown to chemoselectively target tyrosine residues in proteomics experiments, whilst the labelling of histidine was negligible. We hypothesized that sulfonyl-exchange chemistry could be a broadly applicable platform for the specific targeting of histidine residues in proteins through the rational design of chemical probes with optimal equilibrium binding interactions.Cereblon (CRBN) is a component of an E3 ubiquitin ligase complex that was shown to be the physiologically relevant target of immunomodulatory drugs (or IMiDs) such as thalidomide (Fig. 1a).23 IMiDs have been described as ‘molecular glues’24,25 because they bind CRBN and remodel the protein surface resulting in the recruitment of protein neosubstrates, forming ternary complexes that induce polyubiquitination and subsequent proteasomal degradation of the recruited proteins.26 IMiDs have also been incorporated into heterobifunctional molecules called proteolysis targeting chimeras (PROTACs) that co-locate CRBN and target proteins resulting in their polyubiquitination and proteasomal degradation.27 Covalent modulators of CRBN have thus far not been reported in the literature due to the lack of a reactive cysteine residue in the IMiD site. Here, we report the rational targeting of a histidine residue (His353) in the IMiD binding pocket of CRBN that plays a key role in the recruitment of degraded substrates. A fluorosulfate-containing IMiD synthetically re-engineered CRBN, inducing the degradation of a new biological target, while sulfonyl fluoride and sulfonyl triazole probes were shown to be potent CRBN inhibitors that have utility in target validation experiments.
 | ||
| Fig. 1 (a) Chemical structures of the immunomodulatory drugs thalidomide, EM12 and lenalidomide. (b) Crystal structure of the lenalidomide (green), CRBN (grey), DDB1 (cyan) and CK1α (gold) complex (PDB 5FQD).22 (c) Inset shows the proximity of CRBN His353 to the 6-position of lenalidomide. | ||
Results and discussion
The IMiD binding site in CRBN has been well-characterized using X-ray crystallography.28 The histidine residue His353 is proximal not only to the benzene ring of the IMiDs such as lenalidomide (Fig. 1), but it is also close to the recruited neosubstrates of CRBN.22,29,30 We reasoned that covalent engagement of His353 would modulate complex formation leading either to inhibition of the molecular glue degradation mechanism or to the recruitment of alternative neosubstrates compared to reversible binding IMiDs through subtle changes of the protein surface. The crystal structure of the lenalidomide/CRBN/DDB1 complex suggested that incorporation of a sulfonyl fluoride electrophilic warhead at the 6-position of the isoindolinone ring would provide the optimal location for templated covalent adduct formation (Fig. 1b and c). We chose to use the synthetically simpler core structure of EM12 as the focus of our design strategy. Sulfonyl fluoride 1 (EM12-SO2F, Fig. 2) was prepared using a published procedure that utilized a palladium-mediated conversion of 6-bromo-EM12 to the benzyl sulfide, that was subsequently oxidized using N-chlorosuccinimide to the sulfonyl chloride and converted to the desired fluoride using KF (synthetic details are provided for all probes in the ESI†).10 To assess CRBN binding in cells a NanoBRET assay was developed that measures the dose-dependent reduction in BRET signal following displacement of a fluorescent CRBN tracer from NanoLuc-tagged CRBN, similar to a recently reported assay that used transient transfection.31 Interestingly, we found that the previously developed BODIPY-lenalidomide probe32 is cell permeable and results in efficient NanoBRET signal measured by a custom designed 450-80/520 BRET module (BMG Labtech, ESI). EM12-SO2F was found to be an extremely potent ligand of CRBN in cells. Intact mass spectrometry of the recombinant CRBN/DDB1 complex revealed adduct formation as predicted (Fig. 3a and ESI†). A biochemical CRBN TR-FRET binding assay using terbium-CRBN and BODIPY-lenalidomide was developed that confirmed a rapid time-dependent increase in potency of EM12-SO2F (ESI†). The regioisomer 2 (Fig. 2) positions the warhead in a less optimal location, which translates to lower potency as expected, yet the probe is still able to form an adduct with CRBN and labels His353 as shown by peptide mapping mass spectrometry (MS) (see ESI†). Changing the fluorine leaving group to triazoles 3 and 4 (synthetically achieved in one step from the sulfonyl chloride) retained potent inhibition of CRBN, though adduct formation was only observed for probe 3 (labelling of His353 was confirmed using peptide mapping). We surmised that instability of probe 4 may be the reason for the observed lack of labelling, and human plasma stability assessment of the sulfonyl exchange ligands appeared consistent with this observation (Fig. 2). We decided to prepare the intrinsically less electrophilic fluorosulfate warhead33 to assess whether equilibrium binding interactions of the IMiD scaffold were sufficient to template the desired reaction with histidine. Probe 5 (EM12-FS, Fig. 2) was prepared from the precursor phenol using [4-(acetylamino)phenyl]imidodisulfuryl difluoride (AISF), a reagent developed recently for the synthesis of fluorosulfates.34 EM12-FS retained CRBN binding affinity and labelled the protein at His353 exclusively. The regioisomer 6, which was prepared analogously from the phenol and AISF, was found to be a weaker binder than EM12-FS (Fig. 2) and only partially labelled CRBN (ESI†) as expected based on the result of the isomeric sulfonyl fluoride congeners 1 and 2. Isomer 7 engages CRBN quite potently, presumably due to more optimal equilibrium binding interactions with the protein, since there is negligible covalent modification of the protein (ESI†). Further metabolic profiling of EM12-FS revealed impressive stability in human plasma, microsomes (HLM) and hepatocytes (Hhep), in line with its lower intrinsic reactivity compared to the sulfonyl fluoride and sulfonyl triazoles (Fig. 2).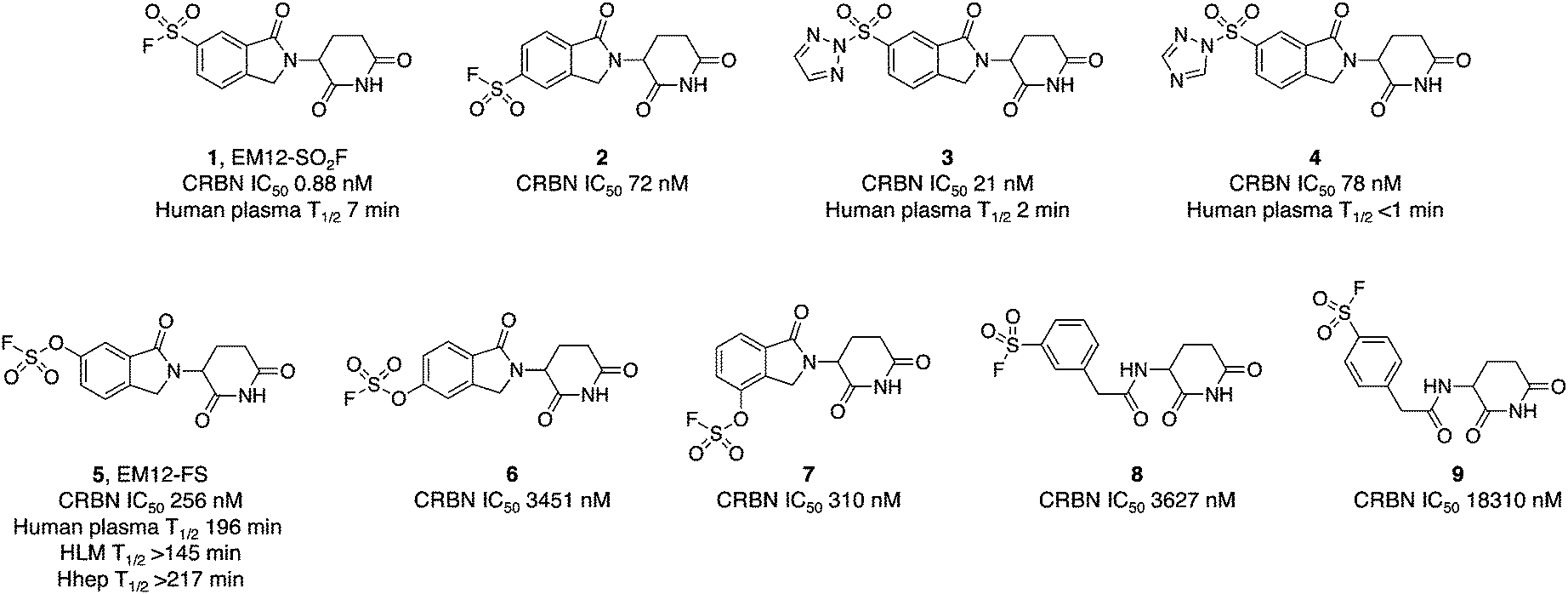 | ||
| Fig. 2 Cellular potency measured in a NanoBRET engagement assay and metabolic stability of sulfonyl exchange probes targeting cereblon. | ||
 | ||
| Fig. 3 (a) Intact MS of CRBN/DDB1 (molecular weight trace of CRBN shown) and the mass shift of CRBN following treatment with 1 eq. EM12-SO2F (4 hours), which is commensurate with sulfonylation (Δmass = 307). (b) EM12-SO2F inhibits the degradation of IKZF1 by lenalidomide (Len) as shown by WB in MOLT4 cells (2 hours pre-treatment with EM12-SO2F followed by 5 hours incubation with lenalidomide). | ||
The human plasma half-life of EM12-FS is superior to that of thalidomide (22 minutes) and EM12 (119 minutes), and between those of pomalidomide and lenalidomide (159 and 284 minutes respectively).35 These results demonstrate that the fluorosulfate warhead possesses adequate stability to be suitable for covalent drug design strategies.36
We also explored structure-activity relationships and CRBN labelling in a simple, previously reported, benzylamide series of CRBN ligands.37 Regioisomeric probes 8 and 9 were weak binders of CRBN and yet still able to covalently engage the protein as shown by intact MS (Fig. 2 and ESI†).
The pharmacological effects of incorporating such covalent warheads into EM12 were determined using quantitative degradation proteomics in MOLT4 cells employing a procedure that was recently published.38 Probes 1–4, 8 and 9 do not degrade a single protein to any considerable extent, likely due to deleterious His353 conformations that perturb productive neosubstrate complex formation (ESI†).
To demonstrate the utility of EM12-SO2F in cell-based target validation experiments, we showed that the degradation of the neosubstrate zinc finger transcription factor IKZF1 by lenalidomide was inhibited in cells by the highly potent sulfonyl fluoride probe (Fig. 3b). EM12-SO2F thus complements CRBN-targeting PROTACs39,40 and genetic methods of perturbation (CRISPR, RNAi) to validate phenotypic modes-of-action that depend on CRBN-mediated degradation.41
However, proteomic profiling of the fluorosulfate EM12-FS in MOLT4 cells revealed selective downregulation of NTAQ1, a protein N-terminal glutamine amidohydrolase involved in the Arg/N-end rule pathway of protein degradation (Fig. 4a).42 Further confirmation was achieved by showing exclusive degradation of NTAQ1 in an additional cell line using quantitative MS proteomics (THP1, see ESI†). NTAQ1 was confirmed as a degraded target in western blot follow-up experiments using FLAG-tagged NTAQ1 (blotting for both anti-NTAQ1 and anti-FLAG, Fig. 4b). The activity of cullin ring ligases (CRLs) has been shown to be reliant upon neddylation, and the NEDD8-activating enzyme (NAE) inhibitor MLN4924 is often used as confirmatory evidence that degradation is mediated by CRLs, including the CRBN E3 ubiquitin ligase complex CRL4CRBN.43,44 Pre-treatment of cells with MLN4924 rescued downregulation of NTAQ1 by EM12-FS, showing that degradation required the active CRL (Fig. 4b).
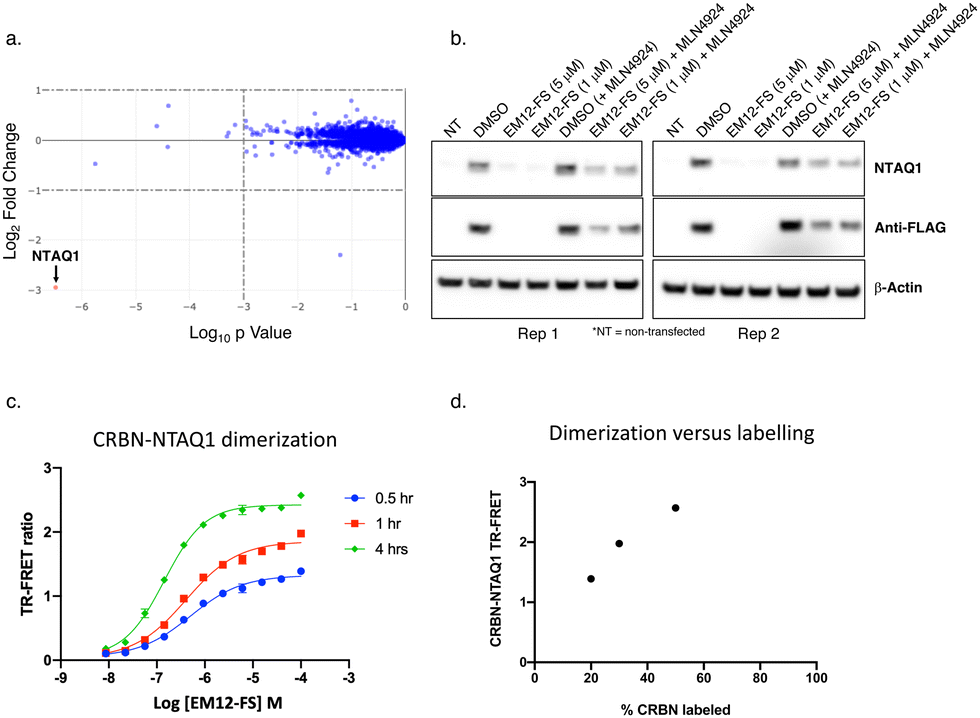 | ||
| Fig. 4 (a) MOLT4 cells were treated with EM12-FS and protein abundance changes were determined using MS proteomics. NTAQ1 was the only protein degraded to a significant extent (red dot). (b) EM12-FS degrades NTAQ1 in MOLT4 cells in a neddylation-dependent manner. (c) Time-dependent dimerization of CRBN and NTAQ1 mediated by EM12-FS measured by TR-FRET. (d) Correlation of CRBN-NTAQ1 maximum TR-FRET ratio from (c) with the extent of CRBN labelling by EM12-FS, determined using intact MS (see ESI†). | ||
A TR-FRET CRBN-NTAQ1 dimerization assay was developed to further validate the molecular mode-of-action of EM12-FS (ESI†). Time-dependent increases in the dimerization of CRBN-NTAQ1 induced by EM12-FS correlated with the extent of CRBN labelling, strongly suggesting that covalent engagement of His353 is required to recruit and degrade NTAQ1 (Fig. 4c and d). The extent of dimerization induced by EM12-SO2F is considerably less than EM12-FS (see ESI†), suggesting that the subtle changes in conformation formed within the complex through sulfonyl exchange chemistry affect ubiquitination and subsequent degradation of NTAQ1.
Conclusions
Sulfonyl exchange chemistry has emerged as a powerful platform to rationally target nucleophilic amino acid residues such as tyrosine, lysine and serine in protein binding sites, delivering covalent chemical probes and functional chemical biology tools. This work shows for the first time that sulfur exchange chemistry is well-suited to the development of histidine targeting small molecule protein ligands and drugs. Synthetic modification of CRBN through rational targeting of a histidine residue in the IMiD binding site using sulfonyl exchange chemistry yielded potent inhibitors and a degrader of a novel neosubstrate. Sulfonyl fluoride and triazole probes covalently modify His353 and do not possess significant neosubstrate degradation capacity. These probes block the IMiD binding site and therefore supplement the CRBN target validation toolkit. Remarkably, the addition of a single oxygen atom to yield the intrinsically less electrophilic fluorosulfate EM12-FS also modified His353 exclusively but induced the efficient heterodimerization of CRBN and NTAQ1 leading to the selective degradation of the latter. This is the first reported example of a small molecule modulator of NTAQ1, a protein annotated as unligandable.45 NTAQ1 is involved in the DNA damage response and the degraders described here may be useful starting points for the development of anticancer agents.46 Our results suggest that binding in the IMiD site and covalent engagement of His353 is not sufficient to trigger degradation of NTAQ1. The subtle variations in conformation at the CRBN surface effected by different electrophilic warheads appear to control the functionality of the re-engineered complex. We expect the sulfonyl oxygen atoms present in the probes described here to clash with the G-loop degron, perhaps explaining why traditional neosubstrates such as zinc finger transcription factors do not appear to be degraded. NTAQ1 possibly presents a new degron motif, although additional biochemical and structural biology studies are required to categorically confirm this. Nevertheless, our work demonstrates that inhibitor EM12-SO2F and degrader EM12-FS are useful chemical probes that will enable further biological studies of CRBN, NTAQ1 and the Arg/N-end rule pathway.A lenalidomide derivative bearing a diazirine photoaffinity handle was recently shown to label His353 in CRBN following UV irradiation,47 through a mechanism that may involve nucleophilic attack of the imidazole residue on the diazo intermediate.48 This study and ours reveal the surprisingly high reactivity of His353 in CRBN (a residue that is highly conserved across species) and we thus speculate that this may reflect its possible role in the endogenous function of CRBN. Previously, sulfonyl exchange chemistry has been shown to identify protein phosphosites and nucleophilic residues involved in (oligo)nucleotide binding/cleavage and further studies may unearth such a role for His353.11,19
Previously, the mode-of-action of the natural products fumagillin and ovalicin were determined to be through fortuitous labelling of a histidine residue in the active site of methionine aminopeptidase 2 (MetAP-2) by their spiroepoxide electrophiles.49,50 Ovalicin was conjugated to an IκBα phosphopeptide that is recognized by the ubiquitin ligase complex SCFβ-TRCP to create the first PROTAC molecule that degraded MetAP-2.51 Having established the feasibility of covalently modifying CRBN, our work also enables the future development of covalent PROTACs but with enhanced pharmacodynamic efficacy and catalytic efficiency because the warhead labels the E3 complex rather than the protein-of-interest.52
Additionally, other programmable E3 ligases possess ligandable residues beyond cysteine (including tyrosine, lysine and now histidine). Covalent remodelling of the surface of E3 complexes could provide a general strategy to explore the recruitment and degradation of diverse neosubstrates. We also believe suitably designed covalent ligands are capable of site-selective chemical mutagenesis in cells enabling synthetic modification of protein surfaces to engender neofunctionalization of the target. Applications of these methods should find broad utility in synthetic biology and drug discovery.
Author contributions
JTC, GPD, SF, ESF, FH, NRK, RPN and BLZ performed and/or designed the biochemical assays; KAD, SBF, JAM and RJM performed the mass spectrometry studies; LHJ, NSG, HL and JC designed the compounds; LHJ conceived and directed the project; LHJ wrote the manuscript with contributions from all authors.Conflicts of interest
L. H. J. serves on the scientific advisory boards for, and holds equity in, Interline Therapeutics and Rapafusyn Pharmaceuticals, consults for Umbra Therapeutics, Ananke Therapeutics and Matchpoint Therapeutics, and holds equity in Jnana Therapeutics. The Center for Protein Degradation at DFCI receives research funding from Deerfield. E. S. F. is a founder, member of the SAB, and equity holder of Civetta Therapeutics, Jengu Therapeutics, Proximity Therapeutics, and Neomorph Inc, SAB member and equity holder in Avilar Therapeutics and Photys Therapeutics, and a consultant to Astellas, Sanofi, Novartis, Deerfield and EcoR1 capital. The Fischer laboratory receives or has received research funding from Novartis, Deerfield, Ajax, Interline, and Astellas. N. S. G. is a founder, an SAB, and an equity holder in Syros, Jengu, Inception, C4, B2S, Voronoi, Matchpoint, GSK, CobroVentures, Larkspur (board member), and Soltego (board member). The Gray laboratory receives or has received research funding from Novartis, Takeda, Astellas, Taiho, Janssen, Kinogen, Voronoi, Arbella, Epiphanes, Deerfield, and Sanofi. J. C. is a consultant for Soltego, Jengu, Allorion and Matchpoint Therapeutics, and holds equity in Soltego, Allorion, Matchpoint and M3 Bioinformatics & Technology Inc. K. A. D. is a consultant for Kronos Bio and Neomorph Inc. J. A. M. serves on the SAB of 908 Devices and receives sponsored research support from Vertex, AstraZeneca, Taiho, and TUO.Acknowledgements
We thank Wuxi and BioDuro for compound synthesis, and for running the metabolic stability and CRBN NanoBRET assays. J. A. M. acknowledges support from the NIH (R21 CA247671 and R01 CA233800).References
- M. Gehringer and S. A. Laufer, J. Med. Chem., 2019, 62, 5673–5724 CrossRef CAS PubMed.
- J. M. Strelow, SLAS Discovery, 2017, 22, 3–20 CrossRef CAS PubMed.
- F. Daryaee, Z. Zhang, K. R. Gogarty, Y. Li, J. Merino, S. L. Fisher and P. J. Tonge, Chem. Sci., 2017, 8, 3434–3443 RSC.
- Q. Liu, Y. Sabnis, Z. Zhao, T. Zhang, S. J. Buhrlage, L. H. Jones and N. S. Gray, Chem. Biol., 2013, 20, 146–159 CrossRef CAS PubMed.
- P. Ábrányi-Balogh, L. Petri, T. Imre, P. Szijj, A. Scarpino, M. Hrast, A. Mitrović, U. P. Fonovič, K. Németh, H. Barreteau, D. I. Roper, K. Horváti, G. G. Ferenczy, J. Kos, J. Ilaš, S. Gobec and G. M. Keserű, Eur. J. Med. Chem., 2018, 160, 94–107 CrossRef PubMed.
- N. A. Khazanov and H. A. Carlson, PLoS Comput. Biol., 2013, 9, e1003321 CrossRef PubMed.
- L. H. Jones, Annu. Rep. Med. Chem., 2021, 56, 95–134 CAS.
- L. H. Jones, Angew. Chem., Int. Ed., 2018, 57, 9220–9223 CrossRef CAS PubMed.
- H. Mukherjee and N. P. Grimster, Curr. Opin. Chem. Biol., 2018, 44, 30–38 CrossRef CAS PubMed.
- J. Dong, L. Krasnova, M. G. Finn and K. B. Sharpless, Angew. Chem., Int. Ed., 2014, 53, 9430–9448 CrossRef CAS PubMed.
- A. Narayanan and L. H. Jones, Chem. Sci., 2015, 6, 2650–2659 RSC.
- L. H. Jones and J. W. Kelly, RSC Med. Chem., 2020, 11, 10–17 RSC.
- A. J. M. Ribeiro, J. D. Tyzack, N. Borkakoti, G. L. Holliday and J. M. Thornton, J. Biol. Chem., 2020, 295, 314–324 CrossRef CAS PubMed.
- D. Corcoran, N. Maltbie, S. Sudalairaj, F. N. Baker, J. Hirschfeld and A. Porollo, Front. Bioinform., 2021, 1, 653681 CrossRef PubMed.
- S. Soga, H. Shirai, M. Kobori and N. Hirayama, J. Chem. Inf. Model., 2007, 47, 400–406 CrossRef CAS PubMed.
- D. A. Bullough and W. S. Allison, J. Biol. Chem., 1986, 261, 5722–5730 CrossRef CAS.
- K. W. Harlow and R. L. Switzer, J. Biol. Chem., 1990, 265, 5487–5493 CrossRef CAS PubMed.
- L. Gambini, C. Baggio, P. Udompholkul, J. Jossart, A. F. Salem, J. J. P. Perry and M. Pellecchia, J. Med. Chem., 2019, 62, 5616–5627 CrossRef CAS PubMed.
- H. S. Hahm, E. K. Toroitich, A. L. Borne, J. W. Brulet, A. H. Libby, K. Yuan, T. B. Ware, R. L. McCloud, A. M. Ciancone and K. L. Hsu, Nat. Chem. Biol., 2020, 16, 150–159 CrossRef CAS PubMed.
- J. W. Brulet, A. L. Borne, K. Yuan, A. H. Libby and K. L. Hsu, J. Am. Chem. Soc., 2020, 142, 8270–8280 CrossRef CAS PubMed.
- A. L. Borne, J. W. Brulet, K. Yuan and K. L. Hsu, RSC Chem. Biol., 2021, 2, 322–337 RSC.
- G. Petzold, E. S. Fischer and N. H. Thomä, Nature, 2016, 532, 127–130 CrossRef CAS PubMed.
- T. Ito, H. Ando, T. Suzuki, T. Ogura, K. Hotta, Y. Imamura, Y. Yamaguchi and H. Handa, Science, 2010, 327, 1345–1350 CrossRef CAS PubMed.
- X. Tan, L. I. Calderon-Villalobos, M. Sharon, C. Zheng, C. V. Robinson, M. Estelle and N. Zheng, Nature, 2007, 446, 640–645 CrossRef CAS PubMed.
- S. L. Schreiber, Cell, 2021, 184, 3–9 CrossRef CAS PubMed.
- P. P. Chamberlain and L. G. Hamann, Nat. Chem. Biol., 2019, 15, 937–944 CrossRef CAS PubMed.
- G. M. Burslem and C. M. Crews, Cell, 2020, 181, 102–114 CrossRef CAS PubMed.
- Z. Kozicka and N. H. Thomä, Cell Chem. Biol., 2021, 28, 1032–1047 CrossRef CAS PubMed.
- P. P. Chamberlain, A. Lopez-Girona, K. Miller, G. Carmel, B. Pagarigan, B. Chie-Leon, E. Rychak, L. G. Corral, Y. J. Ren, M. Wang, M. Riley, S. L. Delker, T. Ito, H. Ando, T. Mori, Y. Hirano, H. Handa, T. Hakoshima, T. O. Daniel and B. E. Cathers, Nat. Struct. Mol. Biol., 2014, 21, 803–809 CrossRef CAS PubMed.
- Q. L. Sievers, G. Petzold, R. D. Bunker, A. Renneville, M. Słabicki, B. J. Liddicoat, W. Abdulrahman, T. Mikkelsen, B. L. Ebert and N. H. Thomä, Science, 2018, 362, eaat0572 CrossRef PubMed.
- J. D. Vasta, C. R. Corona and M. B. Robers, Methods Mol. Biol., 2021, 2365, 265–282 CrossRef PubMed.
- E. S. Wang, A. L. Verano, R. P. Nowak, J. C. Yuan, K. A. Donovan, N. A. Eleuteri, H. Yue, K. H. Ngo, P. H. Lizotte, P. C. Gokhale, N. S. Gray and E. S. Fischer, Nat. Chem. Biol., 2021, 17, 711–717 CrossRef CAS PubMed.
- L. H. Jones, ACS Med. Chem. Lett., 2018, 9, 584–586 CrossRef CAS PubMed.
- H. Zhou, P. Mukherjee, R. Liu, E. Evrard, D. Wang, J. M. Humphrey, T. W. Butler, L. R. Hoth, J. B. Sperry, S. K. Sakata, C. J. Helal and C. W. Am Ende, Org. Lett., 2018, 20, 812–815 CrossRef CAS PubMed.
- N. R. Kong, H. Liu, J. Che and L. H. Jones, ACS Med. Chem. Lett., 2021, 12, 1861–1865 CrossRef CAS PubMed.
- O. O. Fadeyi, L. R. Hoth, C. Choi, X. Feng, A. Gopalsamy, E. C. Hett, R. E. Kyne, R. P. Robinson and L. H. Jones, ACS Chem. Biol., 2017, 12, 2015–2020 CrossRef CAS PubMed.
- A. Goergler, R. Norcross, P. Schmid and F. Dey, Kusznir, n.d., WO2019043214, 2019.
- K. A. Donovan, F. M. Ferguson, J. W. Bushman, N. A. Eleuteri, D. Bhunia, S. Ryu, L. Tan, K. Shi, H. Yue, X. Liu, D. Dobrovolsky, B. Jiang, J. Wang, M. Hao, I. You, M. Teng, Y. Liang, J. Hatcher, Z. Li, T. D. Manz, B. Groendyke, W. Hu, Y. Nam, S. Sengupta, H. Cho, I. Shin, M. P. Agius, I. M. Ghobrial, M. W. Ma, J. Che, S. J. Buhrlage, T. Sim, N. S. Gray and E. S. Fischer, Cell, 2020, 183, 1714–1731 CrossRef CAS PubMed.
- C. E. Powell, G. Du, J. W. Bushman, Z. He, T. Zhang, E. S. Fischer and N. S. Gray, RSC Med. Chem., 2021, 12, 1381–1390 RSC.
- M. Girardini, C. Maniaci, S. J. Hughes, A. Testa and A. Ciulli, Bioorg. Med. Chem., 2019, 27, 2466–2479 CrossRef CAS PubMed.
- A. Lopez-Girona, D. Mendy, T. Ito, K. Miller, A. K. Gandhi, J. Kang, S. Karasawa, G. Carmel, P. Jackson, M. Abbasian, A. Mahmoudi, B. Cathers, E. Rychak, S. Gaidarova, R. Chen, P. H. Schafer, H. Handa, T. O. Daniel, J. F. Evans and R. Chopra, Leukemia, 2012, 26, 2326–2335 CrossRef CAS PubMed.
- H. Wang, K. I. Piatkov, C. S. Brower and A. Varshavsky, Mol. Cell, 2009, 34, 686–695 CrossRef CAS PubMed.
- R. J. Deshaies, E. D. Emberley and A. Saha, Subcell. Biochem., 2010, 54, 41–56 CAS.
- T. A. Soucy, P. G. Smith, M. A. Milhollen, A. J. Berger, J. M. Gavin, S. Adhikari, J. E. Brownell, K. E. Burke, D. P. Cardin, S. Critchley, C. A. Cullis, A. Doucette, J. J. Garnsey, J. L. Gaulin, R. E. Gershman, A. R. Lublinsky, A. McDonald, H. Mizutani, U. Narayanan, E. J. Olhava, S. Peluso, M. Rezaei, M. D. Sintchak, T. Talreja, M. P. Thomas, T. Traore, S. Vyskocil, G. S. Weatherhead, J. Yu, J. Zhang, L. R. Dick, C. F. Claiborne, M. Rolfe, J. B. Bolen and S. P. Langston, Nature, 2009, 458, 732–736 CrossRef CAS PubMed.
- C. Mitsopoulos, P. Di Micco, E. V. Fernandez, D. Dolciami, E. Holt, I. L. Mica, E. A. Coker, J. E. Tym, J. Campbell, K. H. Che, B. Ozer, C. Kannas, A. A. Antolin, P. Workman and B. Al-Lazikani, Nucleic Acids Res., 2021, 49, D1074–D1082 CrossRef CAS PubMed.
- K. I. Piatkov, L. Colnaghi, M. Békés, A. Varshavsky and T. T. Huang, Mol. Cell, 2012, 48, 926–933 CrossRef CAS PubMed.
- Z. Lin, Y. Amako, F. Kabir, H. A. Flaxman, B. Budnik and C. M. Woo, J. Am. Chem. Soc., 2022, 144, 606–614 CrossRef CAS PubMed.
- A. V. West, G. Muncipinto, H. Y. Wu, A. C. Huang, M. T. Labenski, L. H. Jones and C. M. Woo, J. Am. Chem. Soc., 2021, 143, 6691–6700 CrossRef CAS PubMed.
- S. Liu, J. Widom, C. W. Kemp, C. M. Crews and J. Clardy, Science, 1998, 282, 1324–1327 CrossRef CAS PubMed.
- E. C. Griffith, Z. Su, B. E. Turk, S. Chen, Y. H. Chang, Z. Wu, K. Biemann and J. O. Liu, Chem. Biol., 1997, 4, 461–471 CrossRef CAS PubMed.
- K. M. Sakamoto, K. B. Kim, A. Kumagai, F. Mercurio, C. M. Crews and R. J. Deshaies, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 8554–8559 CrossRef CAS PubMed.
- N. P. Grimster, RSC Med. Chem., 2021, 12, 1452–1458 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2cb00078d |
| This journal is © The Royal Society of Chemistry 2022 |