
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA dual-responsive doxorubicin–indoximod conjugate for programmed chemoimmunotherapy†
Zhaoxuan
Yang
,
Jiaqi
Huang
,
Yaying
Lin
,
Xiangjie
Luo
,
Haojin
Lin
,
Hongyu
Lin
* and
Jinhao
Gao
 *
*
Fujian Provincial Key Laboratory of Chemical Biology, The MOE Key Laboratory of Spectrochemical Analysis & Instrumentation, and Department of Chemical Biology, College of Chemistry and Chemical Engineering, Xiamen University, Xiamen 361005, China, China. E-mail: hylin007@xmu.edu.cn; jhgao@xmu.edu.cn
First published on 30th May 2022
Abstract
Herein we report a dual-responsive doxorubicin–indoximod conjugate (DOXIND) for programmed chemoimmunotherapy. This conjugate is able to release doxorubicin and indoximod upon exposure to appropriate stimuli for synergistic chemotherapy and immunotherapy, respectively. We demonstrate its promoting effects on immune response and inhibiting effects on tumor growth through a series of in vitro and in vivo experiments.
 Jinhao Gao | Jinhao Gao obtained his BSc from Nanjing University in 2004 and his PhD from The Hong Kong University of Science and Technology (HKUST) in 2008. He was a postdoctoral fellow at Stanford University from 2008 to 2010. He is now a professor of chemical biology at Xiamen University. He was awarded the National Natural Science Fund for Excellent Young Scholars (2012) and Distinguished Young Scholars (2021). His main research interests include chemical biology, nanochemistry, MRI probes, molecular imaging, and cancer therapy. |
Recently, immunotherapy has undergone rapid development, and has become one of the important pillars for cancer therapy, along with surgery, radiotherapy, and chemotherapy. Several types of immunotherapy have been developed based on various mechanisms, including immune checkpoint blockade,1,2 cancer vaccines,3 therapeutic antibodies,1 chimeric antigen receptor (CAR) T-cell therapy,4 and so on. Impeding immunosuppressive signaling pathways related to cancer is also an interesting and encouraging approach. Among assorted targets in this approach, the indoleamine 2,3-dioxygenase (IDO)-mediated pathway is a promising one.5,6 IDO is often overexpressed in the tumor microenvironment (TME), where it converts L-tryptophan (L-Trp) to L-kynurenine (L-Kyn).7 The depletion of Trp and the accumulation of Kyn results in tumor immunosuppression through the interactions among several signaling pathways. Several IDO-inhibitors, such as 1-methyl-D-tryptophan (indoximod, IND) and epacadostat, have entered clinical trials during the past few years.8 However, the single use of IDO-inhibitors showed rather limited therapeutic effects against cancer.9 For example, epacadostat was relatively weak in the treatment of melanoma patients when administered alone.10 Moreover, this is also a predicament that many attempts using this approach have encountered, although they are targeting other immunosuppressive signaling pathways.11,12
Many efforts have been invested in addressing this problem. Several preclinical studies suggest that combination with established chemotherapy may be an effective strategy, especially for chemotherapy prior to immunotherapy.13–20 The idea behind this strategy is that certain chemotherapeutic agents could induce immunogenic cell death (ICD) of cancer cells, resulting in the release of damage-associated molecular patterns (DAMPs),20–25 which considerably boosts the immune response of the host.21–23 Furthermore, in many cases where peripheral immune cells cannot get access to the tumor, chemotherapeutic agents could deplete or inhibit immunosuppressive cells, and promote the release of immune-stimulatory cytokines and immune cell-recruiting chemokines, which also enhances the immune response of the host.24–26 Therefore, suitable chemotherapeutic agents might strengthen the anticancer effects of IDO inhibitors.27,28 On the other hand, overexpression of IDO has been reported to mediate the resistance of cancer cells to chemotherapy.29,30 Downregulation of IDO has been shown to improve the sensitivity of cancer cells to chemotherapy.31 Besides, an elevated level of cytotoxic T cell infiltration, which has been observed in IDO inhibition-based cancer therapy,32 is indicative of favorable prognosis for chemotherapy.33 As a result, significant potential benefits are expected for executing chemotherapy with IDO inhibition. Recently, a few nanosized systems (including inorganic nanoparticles, nanomicelles, and nanogels) based on a similar approach have shown some promising results.22,34,35 However, several obstacles still need to be circumvented during the clinical translation of these systems, such as homogeneity, pharmacokinetics, and large-scale production. On the contrary, molecular conjugates with precise structures, which are amenable to facile adaptation and manageable synthesis, offer a brighter prospect for chemoimmunotherapy. Collectively, the combination of chemotherapy with an ICD character and immunotherapy that inhibits IDO-mediated signaling pathways might achieve considerable anticancer efficacy.
In light of these considerations, we designed a dual-responsive doxorubicin–indoximod conjugate (DOXIND) for programmed cancer chemoimmunotherapy (Fig. 1). This prodrug is expected to release doxorubicin (DOX) when it reaches the mildly acidic TME. DOX is regarded as one of the best chemotherapeutics for inducing ICD.24,27,36 It kills cancer cells and improves the presentation of TAAs to DCs. Meanwhile, the apoptosis of tumor cells raises the level of caspase 3/7, leading to the liberation of IND, which is a well-known IDO pathway inhibitor that is currently in clinical trials.5 IND inhibits IDO-mediated immunosuppression pathways, which results in the recruitment and activation of naïve T cells and the proliferation of cytotoxic T cells, leading to enhanced immune response against tumor cells. As a result, synergistic combinative chemoimmunotherapy is accomplished. We firstly verified the capability of DOXIND for programmed liberation of DOX and IND by exploring its releasing profiles in the presence of appropriate stimuli. We next evaluated the cytotoxicity of DOXIND against cancer cells and illustrated its capacity for inducing calreticulin (CRT) exposure and alleviating IDO-mediated immunosuppression on the cellular level. Lastly, we demonstrated the efficacy of DOXIND for boosting the immune response against cancer and inhibiting the growth of tumors via a series of in vivo experiments.
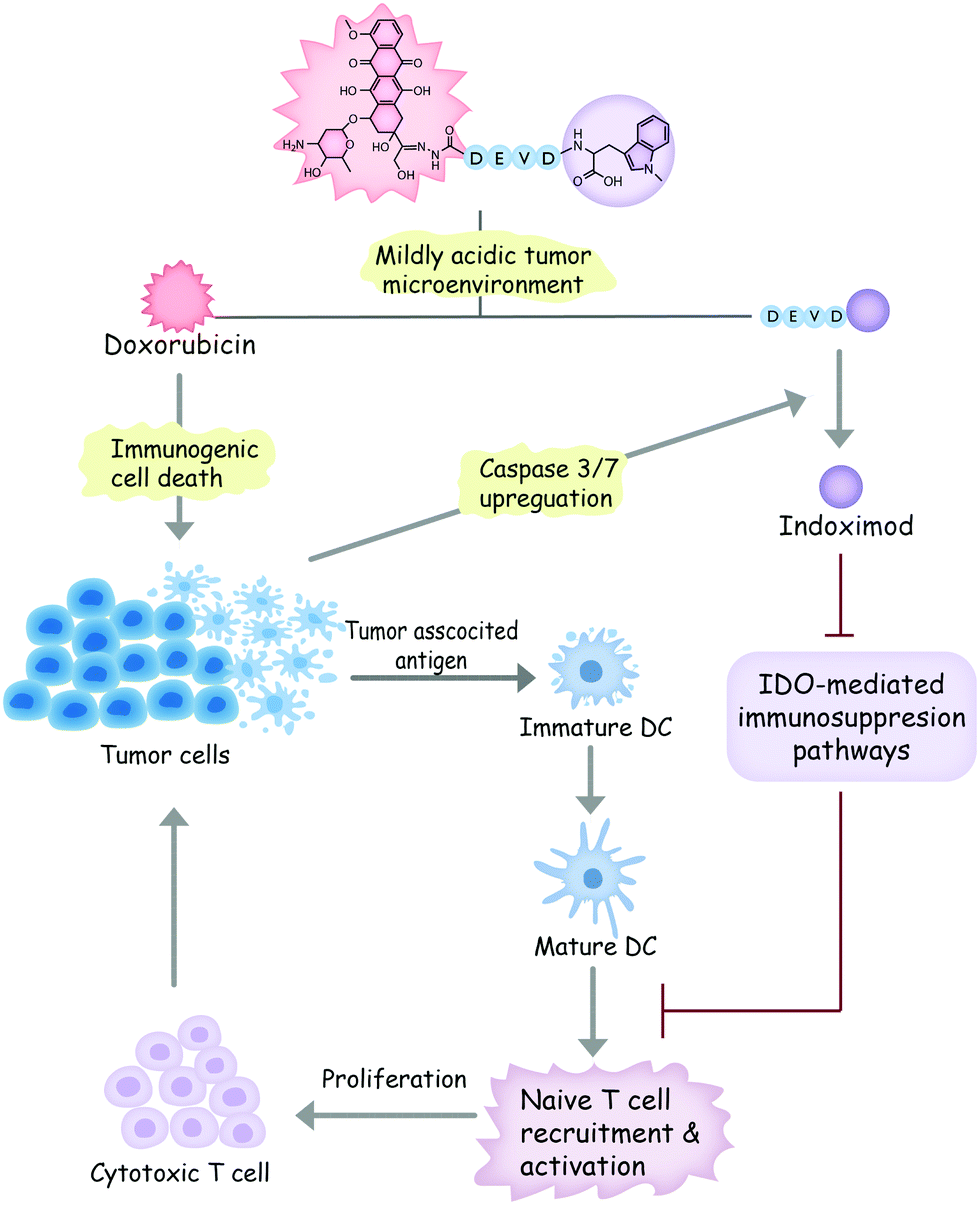 | ||
| Fig. 1 A schematic showing the working mechanism of DOXIND. | ||
DOXIND was synthesized according to Fig. 2. IND (4a) was firstly synthesized from D-tryptophan via methylation. The amino group of IND was then protected to afford Fmoc-IND-OH 3a, which was used as the first amino acid covalently linked to 2-chlorotrityl chloride resins. Fmoc-Asp(OtBu)-OH, Fmoc-Val-OH, Fmoc-Glu(OtBu)-OH, Fmoc-Asp(OtBu)-OH, and Fmoc-NHNHCOCH2CH2-COOH were successively conjugated via standard solid-phase peptide synthesis. After global deprotection, 2a was obtained, which was reacted with DOX to afford DOXIND. The structure of DOXIND was confirmed by high-resolution electrospray ionization mass spectroscopy (HR-ESI-MS) (Fig. S1, ESI†), and the structures of important intermediates were confirmed by nuclear magnetic resonance spectroscopy (NMR) and/or electrospray ionization mass spectrometry (ESI-MS). The hydrazone bond allows for mild acidity-triggered hydrolysis while the DEVD peptide sequence is responsive to caspase 3/7-mediated cleavage, which ensures the programmed release of DOX and IND from DOXIND (Fig. 3a). We quickly tested whether the hydrazine bond could undergo mild acidity-triggered cleavage by high-performance liquid chromatography (HPLC). As shown in Fig. 3b, after the incubation of DOXIND in pH 5.4 PBS buffers, the peak for DOXIND substantially dwindled and a new peak appeared in the HPLC chromatogram, which was confirmed to be DOX, indicating that the hydrazine bond was successfully cleaved under mildly acidic conditions. We further obtained the releasing profiles of DOX from DOXIND under different pH conditions via fluorescence spectroscopy. The presence of IND allows a process called intramolecular photoinduced electron transfer (PET) to occur,37 which significantly attenuates the fluorescence of DOX in DOXIND. The cleavage of the hydrazine bond liberates DOX, resulting in the recovery of DOX fluorescence. This phenomenon was exploited to obtain the release profile of DOX from DOXIND (Fig. S2, ESI†). As shown in Fig. 3c, DOX was quickly discharged from DOXIND and >90% of DOX was released within 4 h under mildly acidic conditions (pH = 5.4). In contrast, only a small amount of DOX (<20%) was liberated under weakly basic conditions (pH = 7.4). We also investigated the release of IND from 2avia HPLC (Fig. 3d). After the incubation of 2a with caspase 3, the peak for 2a diminished and a new peak corresponding to IND appeared in the HPLC chromatogram, which gradually augmented with the extension of the incubation time (Fig. 3e). After 4 h incubation, ∼60% of IND was successful discharged from 2a. These results uncover that DOX and IND could be effectively liberated from DOXIND upon exposure to suitable stimuli as programmed.
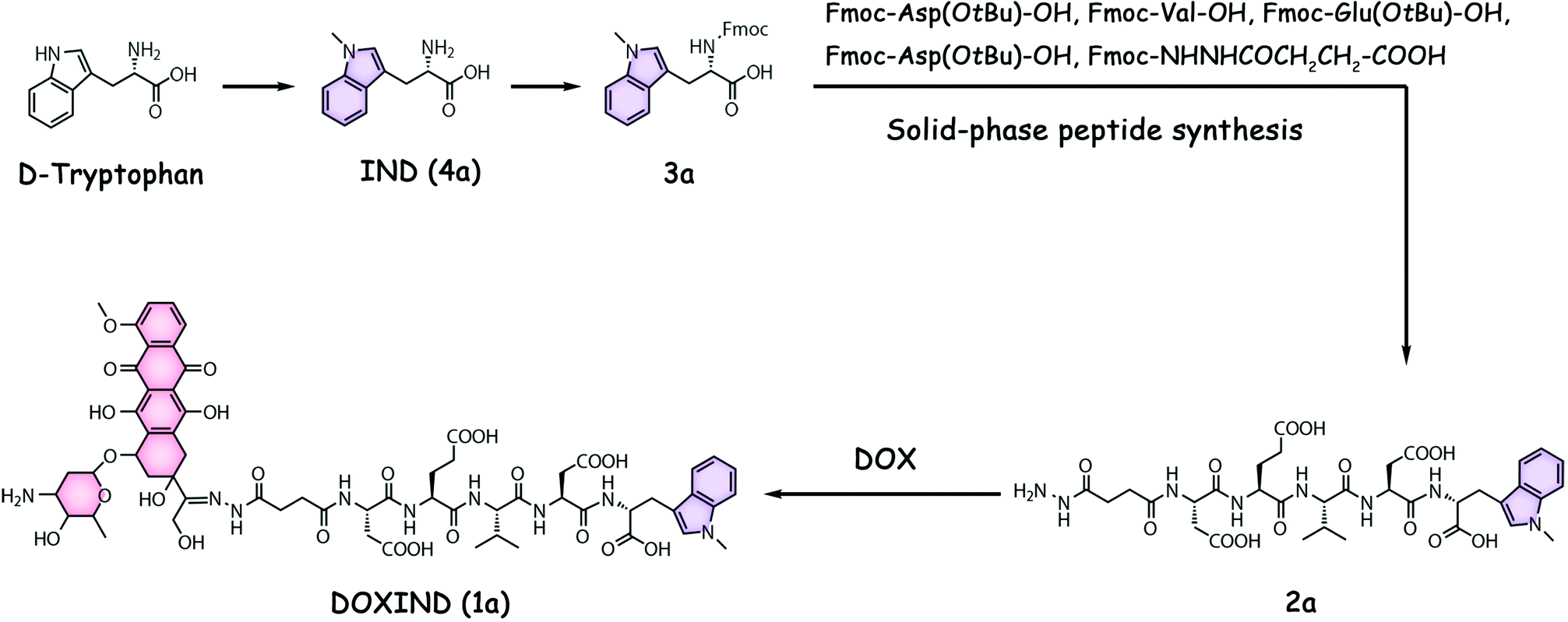 | ||
| Fig. 2 The structure and synthesis of DOXIND. | ||
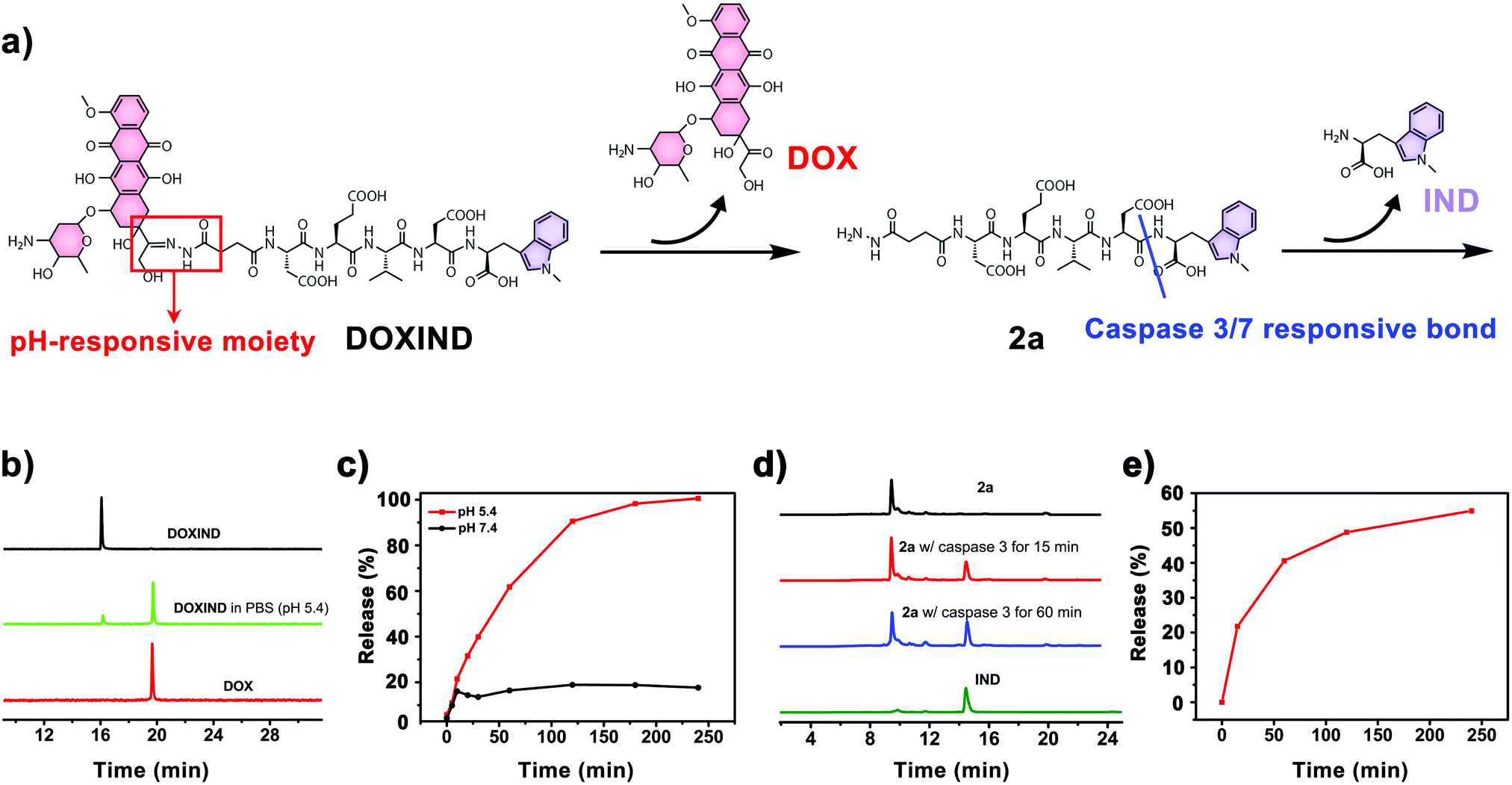 | ||
| Fig. 3 (a) The releasing mechanism of DOX and IND from DOXIND. (b) HPLC chromatograms of DOX and DOXIND (10 μM) in PBS buffers (pH 5.4) for 10 min. (c) The release profile of DOX from DOXIND in PBS buffers (pH 7.4 or 5.4), analyzed by fluorescence spectroscopy. (d) HPLC chromatograms of IND, 2a (2 μM) alone, and 2a incubated with caspase 3 for 15 min or 60 min. (e) The release profile of IND from 2a in the presence of caspase-3, analyzed by HPLC. | ||
We then investigated the anti-cancer performance of DOXIND in cells. We assessed the IC50 of DOXIND and DOX against HeLa and 4T1 cells, which reveals that DOXIND was more cytotoxic than DOX with slightly lower IC50, 0.73 μM and 1.35 μM against HeLa and 4T1 cells, respectively (Fig. 4a and Fig. S3, ESI†). Confocal laser scanning microscopy (CLSM) confirmed the accumulation of released DOX molecules in cell nuclei, where DOX intercalates into DNA and induces cell apoptosis (Fig. S4, ESI†). Evaluation of caspase 3 activity uncovered that the activity of caspase 3 in DOXIND treated HeLa cells was similar to that in DOX treated HeLa cells and much higher than that in PBS treated HeLa cells, allowing for the subsequent liberation of IND from DOXIND (Fig. S5, ESI†). We also studied the level of cell-surface exposure of CRT in treated HeLa cells, which is a well-known indicator of ICD. Flow cytometry discloses that DOXIND could significantly promote the cell-surface exposure of CRT just like DOX, which facilitates downstream elicitation of the immune response (Fig. 4b). We further investigated the effect of DOXIND on activating T cells via mixed leukocyte reaction (MLR) assays. Human peripheral blood mononuclear cells (PBMCs), which are composed of lymphocytes and monocytes, were used to imitate the immune microenvironment. Before being co-cultured with treated HeLa cells, PBMCs were firstly stimulated with a lectin, phytohemagglutinin-M (PHA-M). Carboxyfluorescein succinimidyl ester (CFSE)/CD4+ or CD8+ double-staining flow cytometry was used to evaluate the proliferation of T cells after co-culturing (Fig. S6, ESI†). After co-culturing with HeLa cells, the proliferation rates dropped significantly to 6.82% and 5.73% for CD4+ and CD8+ T cells, respectively (Table 1), which could be attributed to the immunosuppressive pathways that cancer cells activate. This attenuation in T cell proliferation could be considerably relieved by the use of IND during cell co-culturing, which brought the proliferation rates back to 24% and 24.4% for CD4+ and CD8+ T cells, respectively. More importantly, the use of DOXIND during cell co-culturing restored the proliferation rates to 36.3% and 33.8% for CD4+ and CD8+ T cells, respectively. The more prominent rescuing effect of DOXIND could be ascribed to its cytotoxicity against HeLa cells, which also underplays the immunosuppression. Taken together, these results illustrate that DOXIND is capable of annihilating cancer cells, inducing a significant level of CRT exposure, and activating T cells that are subjected to immunosuppressive environments.
 | ||
| Fig. 4 (a) Cytotoxic assessment of DOXIND and DOX against HeLa cells, evaluated with MTT assays. IC50: DOXIND, 0.73 μM; DOX, 0.95 μM. (b) Flow cytometry analysis of CRT exposure in HeLa cells incubated with DOX, DOXIND, or PBS. The treated cells were stained with a primary anti-CRT antibody followed by an Alexa Fluor 680-conjugated secondary antibody. | ||
Lastly, we further evaluated DOXIND's in vivo anti-cancer capacity (Fig. 5). We chose 4T1 tumor-bearing mice because IDO has been reported to be overexpressed in the tumor tissues of 4T1 tumor-bearing mice.38 Overexpression of IDO results in the enhancement of the immunosuppression modulated by intratumoral regulatory T cells (Treg's), leading to poor therapeutic response and accelerated tumor progression.39,40 We therefore investigated the changes of Treg's in the tumor tissues of treated 4T1 tumor-bearing mice. FOXP3 immunohistochemistry (IHC) assays were used to assess the population ratio of Treg's since it is the most commonly-used marker for Treg's.40 As shown in Fig. 5a, the number of FOXP3+ T cells in the tumor tissues of the mice treated with DOXIND is considerably less than that in the tumor tissues of the mice treated with DOX or PBS and comparable to that in the tumor tissues of the mice treated with a combination of DOX and IND, indicating the deactivation of Treg's and the relief of tumor immunosuppression by DOXIND. We also carried out CD8 IHC assays on tumor tissues of treated 4T1 tumor-bearing mice because CD8 is a marker of cytotoxic T cells,41 the infiltration of which into solid tumors is indicative of a positive treatment response and favorable prognosis.42 As shown in Fig. 5b, the infiltrating level of CD8+ T cells in the tumor tissues of the mice treated with DOXIND was significantly higher than those in the tumor tissues of the mice treated otherwise, suggesting the alleviation of tumor immunosuppression and the elicitation of an antitumor immune response by DOXIND. Finally, we evaluated DOXIND's inhibiting effect on tumor growth. 4T1 tumor-bearing mice were randomly separated into four groups and received intravenous injection with PBS, DOXIND, DOX + IND or free DOX, respectively, as indicated in Fig. 5c. The sizes of the tumors were carefully monitored during the treatment period. The average tumor size of the mice treated with DOXIND was substantially smaller than those of the mice treated otherwise, which agrees with the results of IHC assays, demonstrating DOXIND's excellent anticancer efficacy. It is noteworthy that the timing of conducting immunotherapy (after chemotherapy) is critical for achieving the best efficacy of chemoimmunotherapy. Unfortunately, it is challenging to obtain a general guideline for combinational therapy using simply DOX and IND since the best timing varies with different conditions and tumor types. In contrast, the dual-responsive feature of DOXIND ensures sequential on-demand release of doxorubicin and indoximod, which potentially harmonizes the different pharmacokinetics and pharmacodynamics of the two drugs and maximizes the therapeutic efficacy.
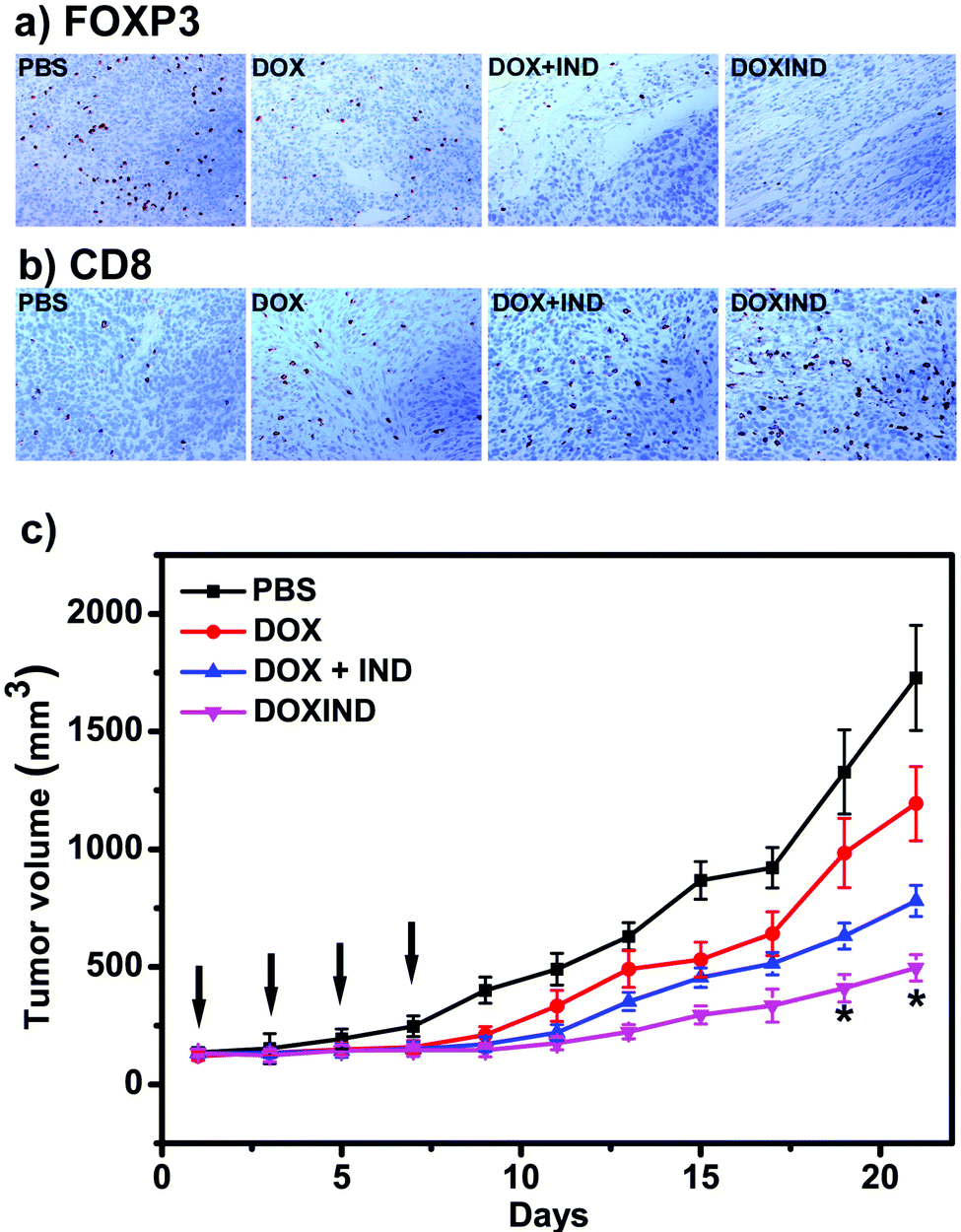 | ||
Fig. 5 (a) IHC staining of FOXP3+ T cells in tumor tissues collected from the mice treated as indicated. See ESI† for details. (b) IHC staining of CD8+ T cells in tumor tissues collected from the mice treated as indicated. See ESI† for details. (c) Tumor growth curves of BALB/c mice bearing 4T1 tumors treated with PBS, DOX, DOX + IND (molar ratio IND![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :DOX = 1:1) or DOXIND at a dosage of 3 mg DOX per kg body weight via tail vein injection every two days for 4 times (indicated by black arrows). The size of each tumor was measured every two days during the treatment. n = 3 for each group, *p < 0.05 compared to the group treated with DOX + IND. :DOX = 1:1) or DOXIND at a dosage of 3 mg DOX per kg body weight via tail vein injection every two days for 4 times (indicated by black arrows). The size of each tumor was measured every two days during the treatment. n = 3 for each group, *p < 0.05 compared to the group treated with DOX + IND. | ||
Conclusions
In conclusion, we developed a dual-responsive DOX-IND conjugate DOXIND for programmed chemoimmunotherapy. This conjugate could discharge DOX in the tumor microenvironment to kill cancer cells and provide an ICD stimulus. In the meantime, the upregulated caspase 3 during the apoptosis of cancer cells could release IND from the residual part of the conjugate, which interferes with the IDO-mediated pathways and alleviates tumor immunosuppression. These effects allow for the elicitation of a significant immune response against cancer cells after the use of ICD-inducing chemotherapeutic agents, which synergistically boosts the potency of both immunotherapy and chemotherapy. We verified the mechanism of action through a series of in vitro and in vivo experiments. Lastly, we illustrated the favorable anticancer efficacy of the conjugate by in vivo treatment experiments, which demonstrates the success of our strategy. More importantly, this strategy could be facilely accommodated for other anticancer drugs that can induce ICD and other inhibiting agents of immunosuppression, which will hopefully stimulate significant innovations in combinative cancer therapy.Animal ethics
All animal procedures were in accordance with the National Institute of Health Guidelines for the care and use of laboratory animals and were approved by the Institutional Animal Care and Use Committee of Xiamen University.Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
The authors acknowledge the research support from the National Natural Science Foundation of China (22125702, 22077107, and 92059109), the Natural Science Foundation of Fujian Province of China (2020J02001), and the Youth Innovation Funding Program of Xiamen City (3502Z20206051).Notes and references
- D. M. Pardoll, Nat. Rev. Cancer, 2012, 12, 252–264 CrossRef CAS PubMed.
- C. Zhang and K. Pu, Chem. Soc. Rev., 2020, 49, 4234–4253 RSC.
- C. J. Melief, T. van Hall, R. Arens, F. Ossendorp and S. H. van der Burg, J. Clin. Invest., 2015, 125, 3401–3412 CrossRef PubMed.
- C. H. June, R. S. O'Connor, O. U. Kawalekar, S. Ghassemi and M. C. Milone, Science, 2018, 359, 1361–1365 CrossRef CAS PubMed.
- S. Lob, A. Konigsrainer, H. G. Rammensee, G. Opelz and P. Terness, Nat. Rev. Cancer, 2009, 9, 445–452 CrossRef PubMed.
- J. Li, C. Xie, J. Huang, Y. Jiang, Q. Miao and K. Pu, Angew. Chem., Int. Ed., 2018, 57, 3995–3998 CrossRef CAS PubMed.
- D. H. Munn and A. L. Mellor, Trends Immunol., 2013, 34, 137–143 CrossRef CAS PubMed.
- S. Qian, M. Zhang, Q. Chen, Y. He, W. Wang and Z. Wang, RSC Adv., 2016, 6, 7575–7581 RSC.
- D. Y. Hou, A. J. Muller, M. D. Sharma, J. DuHadaway, T. Banerjee, M. Johnson, A. L. Mellor, G. C. Prendergast and D. H. Munn, Cancer Res., 2007, 67, 792–801 CrossRef CAS PubMed.
- B. J. Van den Eynde, N. van Baren and J.-F. Baurain, Annu. Rev. Cancer Biol., 2020, 4, 241–256 CrossRef.
- X. Han, K. Cheng, Y. Xu, Y. Wang, H. Min, Y. Zhang, X. Zhao, R. Zhao, G. J. Anderson, L. Ren, G. Nie and Y. Li, J. Am. Chem. Soc., 2020, 142, 2490–2496 CrossRef CAS PubMed.
- K. Cheng, Y. Ding, Y. Zhao, S. Ye, X. Zhao, Y. Zhang, T. Ji, H. Wu, B. Wang, G. J. Anderson, L. Ren and G. Nie, Nano Lett., 2018, 18, 3250–3258 CrossRef CAS PubMed.
- S. J. Antonia, A. Villegas, D. Daniel, D. Vicente, S. Murakami, R. Hui, T. Kurata, A. Chiappori, K. H. Lee, M. de Wit, B. C. Cho, M. Bourhaba, X. Quantin, T. Tokito, T. Mekhail, D. Planchard, Y. C. Kim, C. S. Karapetis, S. Hiret, G. Ostoros, K. Kubota, J. E. Gray, L. Paz-Ares, J. de Castro Carpeno, C. Faivre-Finn, M. Reck, J. Vansteenkiste, D. R. Spigel, C. Wadsworth, G. Melillo, M. Taboada, P. A. Dennis, M. Ozguroglu and P. Investigators, N. Engl. J. Med., 2018, 379, 2342–2350 CrossRef CAS PubMed.
- S. J. Antonia, A. Villegas, D. Daniel, D. Vicente, S. Murakami, R. Hui, T. Yokoi, A. Chiappori, K. H. Lee, M. de Wit, B. C. Cho, M. Bourhaba, X. Quantin, T. Tokito, T. Mekhail, D. Planchard, Y. C. Kim, C. S. Karapetis, S. Hiret, G. Ostoros, K. Kubota, J. E. Gray, L. Paz-Ares, J. de Castro Carpeno, C. Wadsworth, G. Melillo, H. Jiang, Y. Huang, P. A. Dennis, M. Ozguroglu and P. Investigators, N. Engl. J. Med., 2017, 377, 1919–1929 CrossRef CAS PubMed.
- Z. Yang, H. Lin, J. Huang, A. Li, C. Sun, J. Richmond and J. Gao, Chem. Commun., 2019, 55, 4546–4549 RSC.
- J. Xin, K. Zhang, J. Huang, X. Luo, X. Gong, Z. Yang, H. Lin, H. Shan and J. Gao, Biomater. Sci., 2018, 7, 262–271 RSC.
- Q. Chen, L. Xu, C. Liang, C. Wang, R. Peng and Z. Liu, Nat. Commun., 2016, 7, 13193 CrossRef CAS PubMed.
- X. Duan, C. Chan and W. Lin, Angew. Chem., Int. Ed., 2019, 58, 670–680 CrossRef CAS PubMed.
- K. M. Mahoney, P. D. Rennert and G. J. Freeman, Nat. Rev. Drug Discovery, 2015, 14, 561–584 CrossRef CAS PubMed.
- Z. Yang, X. Luo, Y. Lin, J. Huang, H. Lin and J. Gao, ACS Chem. Biol., 2022, 17, 762–767 CrossRef CAS PubMed.
- S. G. Awuah, Y. R. Zheng, P. M. Bruno, M. T. Hemann and S. J. Lippard, J. Am. Chem. Soc., 2015, 137, 14854–14857 CrossRef CAS PubMed.
- J. Lu, X. Liu, Y. P. Liao, F. Salazar, B. Sun, W. Jiang, C. H. Chang, J. Jiang, X. Wang, A. M. Wu, H. Meng and A. E. Nel, Nat. Commun., 2017, 8, 1811 CrossRef PubMed.
- X. Luo, X. Gong, L. Su, H. Lin, Z. Yang, X. Yan and J. Gao, Angew. Chem., Int. Ed., 2021, 60, 1403–1410 CrossRef CAS PubMed.
- J. Wu and D. J. Waxman, Cancer Lett., 2018, 419, 210–221 CrossRef CAS PubMed.
- S. Ladoire, D. Enot, F. Andre, L. Zitvogel and G. Kroemer, Oncoimmunology, 2016, 5, e1082706 CrossRef PubMed.
- L. Bracci, G. Schiavoni, A. Sistigu and F. Belardelli, Cell Death Differ., 2014, 21, 15–25 CrossRef CAS PubMed.
- N. Casares, M. O. Pequignot, A. Tesniere, F. Ghiringhelli, S. Roux, N. Chaput, E. Schmitt, A. Hamai, S. Hervas-Stubbs, M. Obeid, F. Coutant, D. Metivier, E. Pichard, P. Aucouturier, G. Pierron, C. Garrido, L. Zitvogel and G. Kroemer, J. Exp. Med., 2005, 202, 1691–1701 CrossRef CAS PubMed.
- J. Kuang, W. Song, J. Yin, X. Zeng, S. Han, Y.-P. Zhao, J. Tao, C.-J. Liu, X.-H. He and X.-Z. Zhang, Adv. Funct. Mater., 2018, 28, 1800025 CrossRef.
- F. Li, L. Wei, S. Li and J. Liu, Oncotarget, 2017, 8, 107844 CrossRef PubMed.
- S. Maleki Vareki, M. Rytelewski, R. Figueredo, D. Chen, P. J. Ferguson, M. Vincent, W. Min, X. Zheng and J. Koropatnick, Oncotarget, 2014, 5, 2778–2791 CrossRef PubMed.
- S. Maleki Vareki, D. Chen, C. Di Cresce, P. J. Ferguson, R. Figueredo, M. Pampillo, M. Rytelewski, M. Vincent, W. Min, X. Zheng and J. Koropatnick, PLoS One, 2015, 10, e0143435 CrossRef PubMed.
- C. M. Fares, E. M. Van Allen, C. G. Drake, J. P. Allison and S. Hu-Lieskovan, Am. Soc. Clin. Oncol. Educ. Book, 2019, 39, 147–164 CrossRef PubMed.
- Z. Zhang, C. Zhang, G. Zhang, Y. Luo, L. Xue, Q. Zeng, P. Wu, L. Wang, N. Sun and J. He, Clin. Transl. Med., 2021, 11, e456 CAS.
- Q. Song, Y. Yin, L. Shang, T. Wu, D. Zhang, M. Kong, Y. Zhao, Y. He, S. Tan, Y. Guo and Z. Zhang, Nano Lett., 2017, 17, 6366–6375 CrossRef CAS PubMed.
- N. Wang, Z. Wang, Z. Xu, X. Chen and G. Zhu, Angew. Chem., Int. Ed., 2018, 57, 3426–3430 CrossRef CAS PubMed.
- M. Obeid, A. Tesniere, F. Ghiringhelli, G. M. Fimia, L. Apetoh, J. L. Perfettini, M. Castedo, G. Mignot, T. Panaretakis, N. Casares, D. Metivier, N. Larochette, P. van Endert, F. Ciccosanti, M. Piacentini, L. Zitvogel and G. Kroemer, Nat. Med., 2007, 13, 54–61 CrossRef CAS PubMed.
- J. Schubert, A. Schulze, C. Prodromou and H. Neuweiler, Nat. Commun., 2021, 12, 6964 CrossRef CAS PubMed.
- B. Feng, F. Zhou, B. Hou, D. Wang, T. Wang, Y. Fu, Y. Ma, H. Yu and Y. Li, Adv. Mater., 2018, 30, e1803001 CrossRef PubMed.
- Y. W. Moon, J. Hajjar, P. Hwu and A. Naing, J. Immunother. Cancer, 2015, 3, 51 CrossRef PubMed.
- L. Lu, J. Barbi and F. Pan, Nat. Rev. Immunol., 2017, 17, 703–717 CrossRef CAS PubMed.
- J. Galon, A. Costes, F. Sanchez-Cabo, A. Kirilovsky, B. Mlecnik, C. Lagorce-Pages, M. Tosolini, M. Camus, A. Berger, P. Wind, F. Zinzindohoue, P. Bruneval, P. H. Cugnenc, Z. Trajanoski, W. H. Fridman and F. Pages, Science, 2006, 313, 1960–1964 CrossRef CAS PubMed.
- E. Sato, S. H. Olson, J. Ahn, B. Bundy, H. Nishikawa, F. Qian, A. A. Jungbluth, D. Frosina, S. Gnjatic, C. Ambrosone, J. Kepner, T. Odunsi, G. Ritter, S. Lele, Y. T. Chen, H. Ohtani, L. J. Old and K. Odunsi, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 18538–18543 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: The experimental details and Fig. S1–S6. See DOI: https://doi.org/10.1039/d1cb00257k |
| This journal is © The Royal Society of Chemistry 2022 |